
HOMER
Software for motif discovery and ChIP-Seq analysis
Gene Based Analysis (Microarray/RNA-Seq etc.)
There are 3 basic ways to run HOMER - with FASTA files, with Gene
Identifiers, or from Genomic
Positions. This section will outline gene-based
analysis. Gene-based analysis is handled by the
program findMotifs.pl.
This
program
does
more
than
just
finding
motifs, including gene ontology analysis.
By default, this is a promoter-based motif finding analysis,
but can also be used to look for RNA motifs in mRNAs.Input Files:
findMotifs.pl will
analyze the promoters of genes and look for motifs that
are enriched in your target gene promoters relative to
other promoters. The idea is to provide a list of
genes that you believe should contain the same elements,
such as genes that are co-regulated. For example,
you may want to analyze the genes that are up-regulated by
a stimulus, or genes that are specific to a certain cell
type, or genes that appear in the same gene-expression
cluster when doing clustering analysis.
Alternatively, the gene IDs could come from a promoter
ChIP-Chip experiment where each of the promoters are bound
by the same transcription factor.
The primary input data is a list of gene identifiers, placed in single text file where each line contains a gene ID. This can be a gene expression spreadsheet, but findMotifs.pl will expect the first column to contain a gene ID. Below are examples of acceptable input file formats:
The primary input data is a list of gene identifiers, placed in single text file where each line contains a gene ID. This can be a gene expression spreadsheet, but findMotifs.pl will expect the first column to contain a gene ID. Below are examples of acceptable input file formats:
If you're having trouble
with the program, 9 out of 10 times it is due to an
incorrectly formatted input file. If using EXCEL
(especially on the Mac), make sure to save input files as
"Text (Windows)". HOMER will choke on binary XLS
files. HOMER accepts a broad range of different
types of gene identifiers:
- NCBI Entrez Gene IDs
- NCBI Unigene IDs
- NCBI Refseq IDs (mRNA, protein)
- Ensembl Gene IDs
- Gene Symbols (i.e. Official Gene names, like "Nfkb1" )
- popular affymetrix probe IDs (MOE430, U133plus, U95, U75A)
If your favorite ID isn't
listed above, then you will have to covert to one of these
before using HOMER. Your input file can have a mix
of different IDs too. HOMER will let you know how
many of the IDs it was able to "understand", so you can
give it a try.
Running findMotifs.pl
findMotifs.pl takes 3 mandatory arguments:
A gene ID input file, the name of the promoter set (which
is tied to an organism), and an output directory for all
of the output files. For now, HOMER only supports 7
organisms, although you can contact me (cbenner@ucsd.edu)
if you think it would be good to add more. It is
also possible to add
support for an organism and/or a custom promoter set
yourself. For each of these organisms, a
default "Promoter Set" was constructed based on RefSeq
Genes:
findMotifs.pl will produce a number of output files in the "output directory". The primary output will be in HTML files that should be opened with you favorite web browser.
- human (Homo
sapiens)
- mouse (Mus
musculus)
- rat (Rattus
norvegicus)
- fly (Drosophila melanogaster)
- worm (Caenorhabditis elegans)
- zebrafish (Danio rerio)
- yeast (Saccharomyces
cerevisiae)
findMotifs.pl
<inputfile.txt>
<promoter set> <output directory> [options]
i.e. findMotifs.pl
lpsInducedGenes.pl mouse LPSMotifResults/ -start -400
-end 100 -len 8,10 -p 4
This will search for motifs of length 8 and 10 from -400 to +100 relative to the TSS, using 4 threads (i.e. 4 CPUs)
This will search for motifs of length 8 and 10 from -400 to +100 relative to the TSS, using 4 threads (i.e. 4 CPUs)
findMotifs.pl will produce a number of output files in the "output directory". The primary output will be in HTML files that should be opened with you favorite web browser.
What does findMotifs.pl do?
This program performs a
number of operations en route to providing a basic
analysis of motif and functional enrichment. The
various steps are outlined below:
- Convert Gene IDs to consistent gene identifier (usually Entrez Gene ID)
- Select appropriate background IDs (usually all confident genes, i.e. not olfactory genes), or take user supplied list (see below)
- Perform Gene Ontology enrichment calculation (for details, see here).
- Assign weights to background promoters based on the distribution of CpG content in the target gene promoters such that comparable numbers of low and high-CpG promoters are analyzed.
- Perform de novo motif analysis
- Create output HTML pages for de novo analysis.
- Perform known motif enrichment analysis and corresponding output pages.
Output files
The first output page
created is for de novo
results (beside the GO analysis). This page contains
a sorted list of non-redundant motifs ranked by their
enrichment p-values. Below is an example generate
using genes up-regulated after 1 hour of treatment with
LPS in murine macrophages (sample gene list):

findMotifs.pl upLPS.mouse.txt mouse
outputDirectory/ -len 8,10,12
This should produce:
This should produce:
Information is provided for
each motif. One thing I've been debating is whether
or not to include my best guess for the motif identity
("Best Match/Details"). The problem is that people
take this result too literally and it can be a huge
problem and a source of misunderstanding. In many
cases the best match doesn't not look very convincing, but
few people seem to pay attention to that (see below).
More information about HOMER motif finding output
More information about HOMER motif finding output
Important motif finding parameters
Repeat Masked vs. Unmasked
Sequences
Promoter Region ("-start <#>" and "-end <#>", default: -300, 50)
Motif length ("-len <#>" or "-len <#>,<#>,...", default 8,10,12)
Mismatches allowed in global optimization phase ("-mis <#>", default: 2)
Number of CPUs to use ("-p <#>", default 1)
Number of motifs to find ("-S <#>", default 25)
Normalize CpG% content instead of GC% content ("-cpg")
Region level autonormalization ("-nlen <#>", default 3, "-nlen 0" to disable)
Motif level autonormalization (-olen <#>, default 0 i.e. disabled)
User defined background genes ("-bg <file of Gene IDs to use as background>")
Binomial enrichment scoring ("-b")
Find enrichment of individual oligos ("-oligo").
Only search for motifs on + strand ("-norevopp")
Mask motifs ("-mask <motif file>")
Optimize motifs ("-opt <motif file>")
Dump FASTA files ("-dumpFasta")
Removing redundant promoters ("-noredun")
Convert IDs to Human for GO analysis ("-humanGO")
Actually, this usually
doesn't matter that
much. Since HOMER is a differential motif
discovery algorithm, common repeats are usually in both
the target and background sequences. However, it
is not uncommon that a transcription factor binds to a
certain class of repeats, which may cause several large
stretches of similar sequence to be processed, biasing
the results. Usually it's safer to go with the
masked version. To use the unmasked version, use "-nomask".
Promoter Region ("-start <#>" and "-end <#>", default: -300, 50)
Different parts of the
promoter can be used for motif finding. In the
"old days", everyone would search 1kb upstream and look
for motifs there. As it turns out, most of the
action is within 200 bp of the promoter, with the motif
density dropping of considerably after that. The
maximum sizes handled by HOMER are -2000 and 2000.
Motif length ("-len <#>" or "-len <#>,<#>,...", default 8,10,12)
Specifies the length of
motifs to be found. HOMER will find motifs of each
size separately and then combine the results at the
end. The length of time it takes to find motifs
increases greatly with increasing size. In
general, it's best to try out enrichment with shorter
lengths (i.e. less than 15) before trying longer
lengths. Much longer motifs can be found with
HOMER, but it's best to use smaller sets of sequence
when trying to find long motifs (i.e. use "-len 20 -start -150 -end 50"),
otherwise it may take way too long (or take too much
memory). The other trick to reduce the total
resource consumption is to reduce the number of
background sequences.
Mismatches allowed in global optimization phase ("-mis <#>", default: 2)
HOMER looks for promising
candidates by initially checking ordinary oligos for
enrichment, allowing mismatches. The more
mismatches you allow, the more sensitive the algorithm,
particularly for longer motifs. However, this also
slows down the algorithm a bit. If searching for
motifs longer than 12-15 bp, it's best to increase this
value to at least 3 or even 4.
Number of CPUs to use ("-p <#>", default 1)
HOMER is now multicore
compliant. It's not perfectly parallelized,
however, certain types of analysis can benefit. In
general, the longer the length of the motif, the better
the speed-up you'll see.
Number of motifs to find ("-S <#>", default 25)
Specifies the number of
motifs of each length to find. 25 is already quite
a bit. If anything, I'd recommend reducing this
number, particularly for long motifs to reduce the total
execution time.
Normalize CpG% content instead of GC% content ("-cpg")
Consider tying if HOMER is
stuck finding "CGCGCGCG"-like motifs. You can also
play around with disabling GC/CpG normalization ("-noweight").
Region level autonormalization ("-nlen <#>", default 3, "-nlen 0" to disable)
Motif level autonormalization (-olen <#>, default 0 i.e. disabled)
Autonormalization attempts
to remove sequence bias from lower order oligos (1-mers,
2-mers ... up to <#>). Region level
autonormalization, which is for 1/2/3 mers by default,
attempts to normalize background regions by adjusting
their weights. If this isn't getting the job done
(autonormalization is not guaranteed to remove all
sequence bias), you can try the more aggressive motif
level autonormalization (-olen <#>). This performs
the autonormalization routine on the oligo table during
de novo motif discovery. (see here for more info)
User defined background genes ("-bg <file of Gene IDs to use as background>")
By default HOMER uses all
other promoters as the background set. You can
choose a specific set of background promoters by placing
the gene identifiers in a file (just like the target
genes) and using the "-bg
<file>" option. These will still be
normalized for CpG% or GC% content just like normal and
autonormalized unless these options are turned off (i.e.
"-nlen 0 -noweight"). This can be very useful
since HOMER is a differential motif discovery algorithm.
Binomial enrichment scoring ("-b")
By default, findMotifs.pl uses
the hypergeometric distribution to score motifs.
If the set of sequences you are analyzing is very large,
you may want to use the binomial to speed things
up. In general, it is recommended to use the
hypergeometric since it does a better job of describing
biological enrichment.
Find enrichment of individual oligos ("-oligo").
This creates output files
in the output directory named oligo.length.txt.
Only search for motifs on + strand ("-norevopp")
By default, HOMER looks
for transcription factor-like motifs on both
strands. This will force it to only look at the +
strand (relative to the TSS, so - strand if the TSS is
on the - strand).
Mask motifs ("-mask <motif file>")
Mask the motif(s) in the
supplied motif file before starting motif finding.
Multiple motifs can be in the motif file.
Optimize motifs ("-opt <motif file>")
Instead of looking for
novel de novo motifs, HOMER will instead try to optimize
the motif supplied. This is cool when trying to
change the length of a motif, or find a very long
version of a given motif. For example, if you
specify "-opt <file>" and "-len 50", it will try
to expand the motif to 50bp and optimize it.
Dump FASTA files ("-dumpFasta")
Like the fact that HOMER
organizes and extracts your sequence files, but don't
care for HOMER as a motif finding algorithm?
That's cool, just specify "-dumpFasta" and the files
"target.fa" and "background.fa" will show up in your
output directory. You can then use them with MEME
or whatever. Just remember, Chuck knows where you
live...
Removing redundant promoters ("-noredun")
By default, HOMER only
keeps one promoter if it is shared by two genes (i.e.
bidirectional promoter) so that the sequence isn't
duplicated. If the duplicated promoter is found in
both the target promoter group and the background group,
the background instance is removed.
Convert IDs to Human for GO analysis ("-humanGO")
Finding Instances of Specific Motifs
By default, HOMER does not
return the locations of each motif found in the motif
discovery process. To recover the motif locations,
you must first select the motifs you're interested in by
getting the "motif file" output by HOMER. You can
combine multiple motifs in single file if you like to form
a "motif library". To identify motif locations, you
have two options:
1. Run findMotifs.pl with the "-find <motif file>" option. This will output a tab-delimited text file with each line containing an instance of the motif in the target peaks. The output is sent to stdout.
1. Run findMotifs.pl with the "-find <motif file>" option. This will output a tab-delimited text file with each line containing an instance of the motif in the target peaks. The output is sent to stdout.
For example: findMotifs.pl lpsGenes.txt
mouse MotifOutputDirectory/ -find motif1.motif >
outputfile.txt
The output file will contain the columns:
The output file will contain the columns:
- Peak/Region ID
- Offset from the TSS
- Sequence of the site
- Name of the Motif
- Strand
- Motif Score (log odds score of the motif matrix,
higher scores are better matches)
2. Run annotatePeaks.pl with
the "-m <motif
file>" option in tss mode (see the here for more
info). To use this option, you must install the proper
genome. Chuck prefers doing it this way.
This will output a tab-delimited text file with each line
containing a peak/region and a column containing instance
of each motif separated by commas to stdout
For example: annotatePeaks.pl tss mm9
-size -300,50 -m motif1.motif > outputfile.txt
The output file will contain columns:
The output file will contain columns:
- Peak/Region ID
- Chromosome
- Start of TSS region
- End of TSS region
- Strand of Peaks
6-18:
annotation information
19. CpG%
20. GC%
21. Motif Instances
...
Motif Instances have the following format:
<distance from TSS>(<sequence>,<strand>,<conservation>)
i.e -29(TAAATCAACA,+,0.00)
To limit the search to only the target set of genes (or any subset of genes), use the option "-list <gene id file>".
This can also be used to find histograms of motif density relative to the TSS - just add the "-hist <#>" option.
19. CpG%
20. GC%
21. Motif Instances
...
Motif Instances have the following format:
<distance from TSS>(<sequence>,<strand>,<conservation>)
i.e -29(TAAATCAACA,+,0.00)
To limit the search to only the target set of genes (or any subset of genes), use the option "-list <gene id file>".
This can also be used to find histograms of motif density relative to the TSS - just add the "-hist <#>" option.
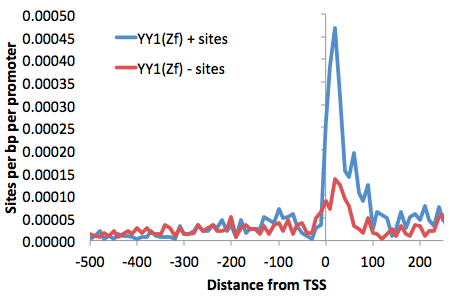
For example: annotatePeaks.pl tss mm9
-size -500,250 -hist 10 -m yy1.motif >
outputfile.txt
Graphing the output file with EXCEL, we can see the distribution of TSS-associated motif YY1:
Add "-list <gene id
list>" to make a histogram on a specific
subset of genes.
Graphing the output file with EXCEL, we can see the distribution of TSS-associated motif YY1:
Practical Tips for Motif Finding

Can't figure something out? Questions, comments, concerns, or other feedback:
cbenner@ucsd.edu