
HOMER
Software for motif discovery and ChIP-Seq analysis
Functional Enrichment Analysis
Below describes HOMER's internal GO calculations which are
provided for convenience. However, if you want to
analyze a gene list of interest consider checking out
Metascape (http://metascape.org/),
which is a full service web application for analyzing one or
more gene lists.HOMER contains a program for performing functional enrichment analysis from a list of Entrez Gene IDs (findGO.pl). Normally, you don't need to know how to use findGO.pl because it is called internally by findMotifs.pl and annotatePeaks.pl. findGO.pl assesses the enrichment of various categories of gene function, biological pathways, domain structure, chromosome location, etc., in your gene list relative to a set of background gene IDs. Enrichment is calculated assuming the cumulative hypergeometric distribution, much in the same way that HOMER scores motif enrichment. HOMER does not attempt to deal with the multiple-hypothesis testing problem, although it does record the number tests made in each output file.
There are several different "ontologies", or libraries of gene groupings, that HOMER will check for enrichment. Below is the list, sources in "()"s:
- Biological Process: Functional groupings of proteins (Gene Ontology)
- Molecular Function: Mechanistic actions of proteins (Gene Ontology)
- Cellular Component: Protein localization (Gene Ontology)
- Chromosome Location: Genes with similar chromosome
localization (NCBI Entrez Gene)
- KEGG Pathways: Groups of proteins in the same pathways (From KEGG)
- Protein-Protein Interactions: Groups of proteins interacting with the same protein (From NCBI Entrez Gene)
- Interpro: Proteins with similar domains and features (Interpro)
- Pfam: Proteins with similar domains and features (Pfam)
- SMART: Proteins with similar domains and features (SMART)
- Gene3D: Proteins with similar domains and features (Gene3D Database)
- Prosite: Proteins with similar domains and features (Prosite Database)
- PRINTS: Proteins with similar domains and features (PRINTS Database)
- MSigDB: Lists of genes maintained by the Molecular Signature Database (includes many different categories of genes (MSigDB)
- BIOCYC: Groups of proteins in the same pathway (NCBI Biosystems/BIOCYC)
- COSMIC: Human proteins that are mutated in the same cancers (COSMIC)
- GWAS Catalog: Human genes with risk SNPs identified in their vicinity for the same disease (GWAS Catalog)
- Lipid Maps: Mouse proteins found in the same lipid processing pathways (NCBI Biosystems/LIPID MAPS)
- Pathway Interaction Database: Proteins in the same pathway (NCBI Biosystems/PID)
- REACTOME: Proteins in the same biochemical pathways (NCBI Biosystems/REACTOME)
- SMPDB: Proteins in the same pathway (SMPDB)
- Wikipathways: Protein in the same pathway
(Wikipathways)
To run findGO.pl on its own, type:
findGO.pl <input file of Entrez Gene
IDs> <organism> <output directory>
[-bg <background ID file>] [-cpu #] [-human]
There are a couple of newer options for findGO.pl that can also be triggered through findMotifs.pl:
-bg <background Gene File> : By default HOMER will use the *.base.gene file found in the homer/data/promoters/ directory for background, which normally represents all gene IDs for the organism. You can use this option to specify a specific background.
-cpu <#> : number of CPUs/threads to use for GO analysis (each ontology will be given it's own thread)
-human : Use Homologene to first convert IDs to human (can useful for non-model organisms) - only way to check COSMIC/GWAS groups if in another organism.
Normally findGO.pl will use a default set of gene ids for that organism. The program produces one HTML file, containing a mixture of different enriched categories, as well tab-delimited text files for each of the ontologies analyzed. An example of the GO output is show below:
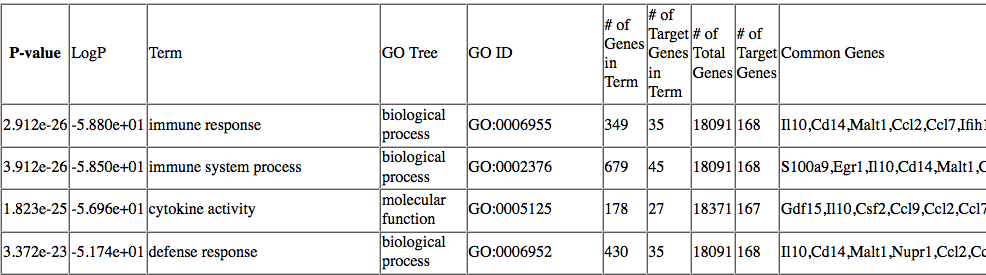
In the HTML page, findGO.pl
will convert the gene IDs into gene symbols so that it is
easy to read. In the text files the IDs are kept as
gene IDs.
Gene Ontology and GO slims
My favorite topic in the
world of Gene Ontology analysis is the use of GO slims.
HOMER does not contain GO slims libraries. As a
result, you may find that many of your gene ontology
results contain terms such as "metabolism" and "cellular process"
when other tools may not reveal these terms. GO
slims are great because they delete terms that you don't
generally want to see. Another way to do this is to
look through your list and just use the terms you
want. There really isn't much of a difference
between that and using GO slims - but at least you're
being honest with yourself with one of the techniques.

Can't figure something out? Questions, comments, concerns, or other feedback:
cbenner@ucsd.edu