
HOMER
Software for motif discovery and next-gen sequencing analysis
Finding Enriched Motifs in Genomic Regions (findMotifsGenome.pl)
HOMER was initially developed to automate the process of
finding enriched motifs in ChIP-Seq peaks. More
generally, HOMER analyzes genomic positions, not limited to
only ChIP-Seq peaks, for enriched motifs. The main
idea is that all the user really needs is a file containing
genomic coordinates (i.e. a HOMER peak file or BED
file), and HOMER will generally take care of the rest.
To analyze a peak file for motifs, run the following
command:findMotifsGenome.pl <peak/BED file>
<genome> <output directory> -size #
[options]
i.e. findMotifsGenome.pl ERpeaks.txt hg18 ER_MotifOutput/ -size 200 -mask
i.e. findMotifsGenome.pl ERpeaks.txt hg18 ER_MotifOutput/ -size 200 -mask
A variety of output files will be placed in the <output directory>, including html pages showing the results. The "-mask" is optional and tells the program to use the repeat-masked sequence. (The old shorthand hg18r will also work). The -size parameter is now mandatory when running findMotifsGenome.pl to avoid confusion - plus it's always a good idea to know exactly what size the regions you are analyzing are.
The findMotifsGenome.pl program is a wrapper that helps set up the data for analysis using the HOMER motif discovery algorithm. By default this will perform de novo motif discovery as well as check the enrichment of known motifs. If you have not done so already, please look over this page describing how HOMER analyzes sequences for enriched motifs.
An important prerequisite for analyzing genomic motifs is that the appropriate genome must by configured for use with HOMER. In version v3.1, HOMER now handles custom/arbitrary genomes. Instead of intalling/configuring a genome, you can specify the path to a file or directory containing the genomic sequence in FASTA format. The genome can be in a single FASTA file, or you specify a directory where where each chromosome can be in a separate file (named chrXXX.fa or chrXXX.fa.masked). In either case, the FASTA headers must contain the chromosome names followed by white space, i.e. ">chr blahblahblah", not ">chr1-blahblahblah", or prefereably only ">chr1". (also note that homer will create a "preparsed/" directory where the genome is, so make sure you have write permissions in the genomic directory.
Selecting the size of the region for motif finding
(-size # or -size given, default: 200)
This is one of the most important parameters and also a source of confusion for many. If you wish to find motifs using your peaks using their exact sizes, use the option "-size given"). However, for Transcription Factor peaks, most of the motifs are found +/- 50-75 bp from the peak center, making it better to use a fixed size rather than depend on your peak size.
Acceptable Input files
findMotifsGenome.pl accepts HOMER peak files
or BED files:
HOMER peak files should have at minimum 5 columns (separated by TABs, additional columns will be ignored):
Mac Users: If using a EXCEL to prepare input files, make sure to save files as a "Text (Windows)" if running MacOS - saving as "Tab delimited text" in Mac produces problems for the software. Otherwise, you can run the script "changeNewLine.pl <filename>" to convert the Mac-formatted text file to a Windows/Dos/Unix formatted text file.
If errors occur, it is likely that the file is not in the correct format, or the first column is not actually populated with unique identifiers.
HOMER peak files should have at minimum 5 columns (separated by TABs, additional columns will be ignored):
- Column1: Unique Peak ID
- Column2: chromosome
- Column3: starting position
- Column4: ending position
- Column5: Strand (+/- or 0/1, where 0="+", 1="-")
- Column1: chromosome
- Column2: starting position
- Column3: ending position
- Column4: Unique Peak ID
- Column5: not used
- Column6: Strand (+/- or 0/1, where 0="+", 1="-")
Mac Users: If using a EXCEL to prepare input files, make sure to save files as a "Text (Windows)" if running MacOS - saving as "Tab delimited text" in Mac produces problems for the software. Otherwise, you can run the script "changeNewLine.pl <filename>" to convert the Mac-formatted text file to a Windows/Dos/Unix formatted text file.
If errors occur, it is likely that the file is not in the correct format, or the first column is not actually populated with unique identifiers.
Custom Background Regions
Since HOMER uses a
differential motif discovery algorithm, different types of
background sequences can be chosen to produce different
results. For example, you may want to compare the
ChIP-Seq peaks specific in one cell type versus the peaks
that are specific to another. To do this, create a
second peak/BED file and use it with the argument "-bg <peak/BED file>".
HOMER
will still try to normalize the background to remove
GC-bias and will also perform autonormalization (see
below). You can turn off the normalization with ("-noweight" and/or "-nlen 0").
Using HOMER2 Background regions
When performing DNA motif discovery with findMotifsGenome.pl, you can add "-useNewBg" to your command to envoke the new positional dependent background correction explained here. This will give you many additional option and control for specifying background sequences, although for many applications the differences may be somewhat minor.
Note: You can always generate background sequences using "homer2 background" and have complete control and then use the resulting FASTA files for motif finding too.
How findMotifsGenome.pl works
There are a series of steps
that the program goes through to find quality motifs:
1. Verify peak/BED file
2. Extract sequences from the genome corresponding to the regions in the input file, filtering sequences that are >70% "N"
3. Calculate GC/CpG content of peak sequences.
4. Preparse the genomic sequences of the selected size to serve as background sequences.
5. Randomly select background regions for motif discovery.
6. Autonormalization of sequence bias.
7. Check enrichment of known motifs
8. de novo motif finding
1. Verify peak/BED file
HOMER makes sure you have
valid peaks, and checks to make sure you have unique
peak identifiers. If there are replicates, it will
inform you, and will add numbers to peak names to ensure
they are unique for downstream analysis.
2. Extract sequences from the genome corresponding to the regions in the input file, filtering sequences that are >70% "N"
This step is pretty self
explanatory. If you wish to extract sequences from
a genome for any reason, check out homerTools.
HOMER will also trash sequences that are predominately
"N". If you feel you are throwing away too many
sequences, try running findMotifsGenome.pl
on an unmasked genome.
3. Calculate GC/CpG content of peak sequences.
CpG Islands are the single
biggest source of sequence content bias in mammalian
genomes, and are unfortunately found near transcription
start sites, where all the action is! By default,
HOMER tracks GC% (use "-cpg"
to use CpG%).
4. Preparse the genomic sequences of the selected size to serve as background sequences.
This step is only done the
first time you find motifs from regions of a given size
("-size <#>").
HOMER takes regions near the TSS of genes (+/- 50kb) and
splits them into regions of the indicated size. It
then calculates their GC/CpG% and stores them for later
use to speed up execution the next time you search for
motifs from similar sized regions.
5. Randomly select background regions for motif discovery.
Since HOMER is a
differential motif discovery algorithm, it must use
background sequence regions as a control. By
default, HOMER selects enough random background regions
such that the total number of regions is 50000 or 2x the
total number of peaks, which ever is larger (to change
use "-N <#>").
The more total sequence that is used, the slower the
program will run, but you want to make sure there is
enough background regions to reliably estimate motif
frequency. HOMER attempts to select background
regions that match the GC-content distribution of the
input sequences (in 5% increments). For example,
if your input regions are extremely GC-rich, HOMER will
select random regions from GC-rich regions of the genome
as a control.
If custom background regions are provided ("-bg <peak/BED file>"), HOMER will automatically ensure that these regions do NOT overlap with the target regions (using mergePeaks). Custom regions will still be normalized for GC-content.
If custom background regions are provided ("-bg <peak/BED file>"), HOMER will automatically ensure that these regions do NOT overlap with the target regions (using mergePeaks). Custom regions will still be normalized for GC-content.
6. Autonormalization of sequence bias.
Autonormalization is a
unique procedure provided by HOMER that attempts to
remove bias introduced by lower-order oligo
sequences. It works by assuming your targets
regions and background regions should not have an
imbalance in 1-mers, 2-mers, 3-mers, etc. The
maximum length of oligo that is autonormalized is
specified by "-nlen
<#>" (default is 3, to disable use "-nlen 0"). For
example, there should not be significantly more A's in
the target sequences that in the background. After
calculating the imbalances for each oligo, it adjusts
the weights of each background sequence by a small
amount to help normalize any imbalance. If target
sequences are rich in A, then background sequences that
contain many A's will be assigned higher weights while
those with very few A's will be assigned lower
weights. The weights are incremented by only small
amounts and the procedure repeated many times in a hill
climbing optimization. This procedure helps remove
some of the sequence bias associated with certain
genomic regions, or bias that may have been introduced
by biased experimental results such as biased
sequencing.
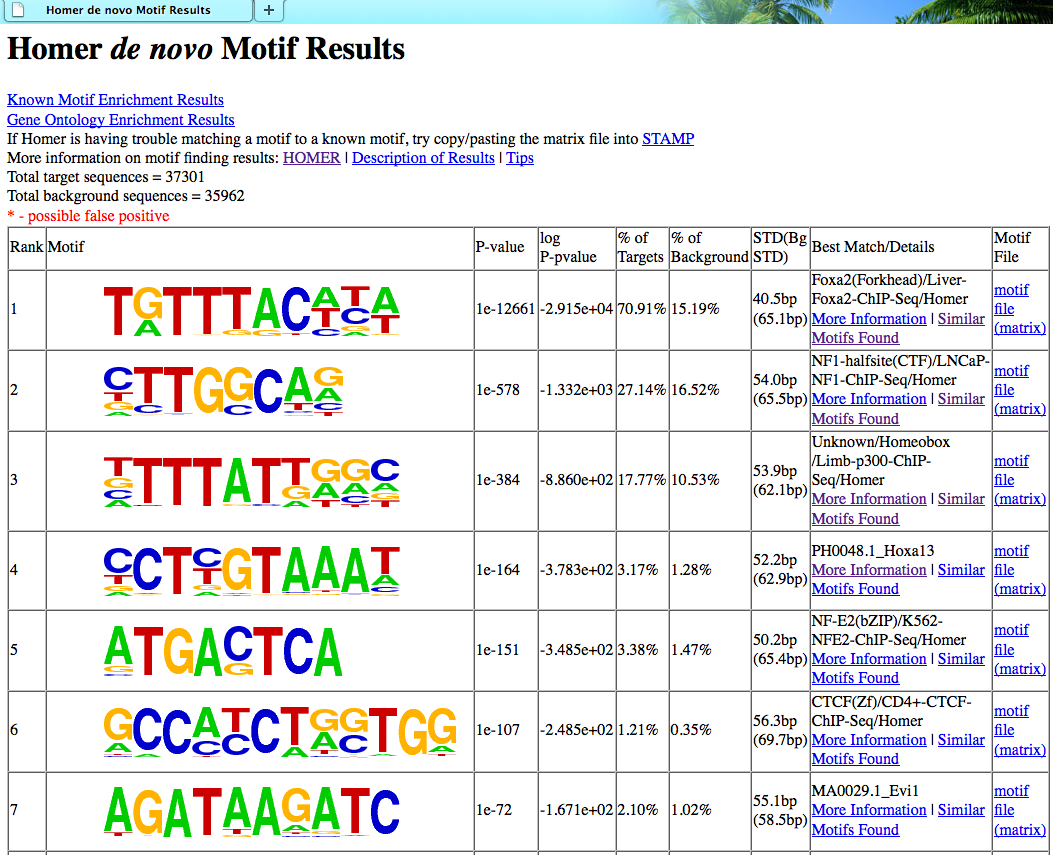
7. Check enrichment of known motifs
HOMER screens it's library
of reliable motifs against the target and background
sequences for enrichment, returning motifs enriched with
a p-value less than 0.05. The known motif
enrichment is performed first since it is usually
faster, and gives a faster look at what's enriched in
your target regions. Know motif enrichment will be
reported to the "knownResults.html" file in the output
directory.
8. de novo motif finding
Best saved for last.
By default, HOMER will search for motifs of len 8, 10,
and 12 bp (change using -len <#,#,#> with no spaces
between the numbers, i.e. "-len 6,10,15,20"). For a more
detail description of the motif discovery algorithm, see
here. Output from the de novo motif finding will
be displayed in the "homerResults.html" file.
findMotifsGenome.pl Output
A full description of motif
finding output and the output can be found here.
Several files are produced in the output directory:
Several files are produced in the output directory:
homerMotifs.motifs<#>
: these are the output files from the de novo motif
finding, separated by motif length, and represent
separate runs of the algorithm.
homerMotifs.all.motifs : Simply the concatenated file composed of all the homerMotifs.motifs<#> files.
motifFindingParameters.txt : command used to execute findMotifsGenome.pl
knownResults.txt : text file containing statistics about known motif enrichment (open in EXCEL).
seq.autonorm.tsv : autonormalization statistics for lower-order oligo normalization.
homerResults.html : formatted output of de novo motif finding.
homerResults/ directory: contains files for the homerResults.html webpage, including motif<#>.motif files for use in finding specific instance of each motif.
knownResults.html : formatted output of known motif finding.
knownResults/ directory: contains files for the knownResults.html webpage, including known<#>.motif files for use in finding specific instance of each motif.
homerMotifs.all.motifs : Simply the concatenated file composed of all the homerMotifs.motifs<#> files.
motifFindingParameters.txt : command used to execute findMotifsGenome.pl
knownResults.txt : text file containing statistics about known motif enrichment (open in EXCEL).
seq.autonorm.tsv : autonormalization statistics for lower-order oligo normalization.
homerResults.html : formatted output of de novo motif finding.
homerResults/ directory: contains files for the homerResults.html webpage, including motif<#>.motif files for use in finding specific instance of each motif.
knownResults.html : formatted output of known motif finding.
knownResults/ directory: contains files for the knownResults.html webpage, including known<#>.motif files for use in finding specific instance of each motif.
Interpreting motif finding results
The format of the output
files generated by findMotifsGenome.pl
are identical to those generated by the promoter-based
version findMotifs.pl
(description).
In general, when analyzing ChIP-Seq / ChIP-Chip peaks you should expect to see strong enrichment for a motif resembling the site recognized by the DNA binding domain of the factor you are studying. Enrichment p-values reported by HOMER should be very very significant (i.e. << 1e-50). If this is not the case, there is a strong possibility that the experiment may have failed in one way or another. For example, the peaks could be of low quality because the factor is not expressed very high.
Practical Tips for Motif finding
In general, when analyzing ChIP-Seq / ChIP-Chip peaks you should expect to see strong enrichment for a motif resembling the site recognized by the DNA binding domain of the factor you are studying. Enrichment p-values reported by HOMER should be very very significant (i.e. << 1e-50). If this is not the case, there is a strong possibility that the experiment may have failed in one way or another. For example, the peaks could be of low quality because the factor is not expressed very high.
Practical Tips for Motif finding
Important motif finding parameters
Masked vs. Unmasked Genome
("-mask" or hg18 vs. hg18r)
Region Size ("-size <#>", "-size <#>,<#>", "-size given", default: 200)
Motif length ("-len <#>" or "-len <#>,<#>,...", default 8,10,12)
Mismatches allowed in global optimization phase ("-mis <#>", default: 2)
Number of CPUs to use ("-p <#>", default 1)
Number of motifs to find ("-S <#>", default 25)
Normalize CpG% content instead of GC% content ("-cpg")
Region level autonormalization ("-nlen <#>", default 3, "-nlen 0" to disable)
Motif level autonormalization (-olen <#>, default 0 i.e. disabled)
User defined background regions ("-bg <peak file of background regions>")
Hypergeometric enrichment scoring ("-h")
Find enrichment of individual oligos ("-oligo").
Force findMotifsGenome.pl to re-preparse genome for the given region size ("-preparse").
Only search for motifs on + strand ("-norevopp")
Search for RNA motifs ("-rna")
Mask motifs ("-mask <motif file>")
Optimize motifs ("-opt <motif file>")
Dump FASTA files ("-dumpFasta")
Actually, this usually
doesn't matter that
much. Since HOMER is a differential motif
discovery algorithm, common repeats are usually in both
the target and background sequences. However, it
is not uncommon that a transcription factor binds to a
certain class of repeats, which may cause several large
stretches of similar sequence to be processed, biasing
the results. Usually it's safer to go with the
masked version.
Region Size ("-size <#>", "-size <#>,<#>", "-size given", default: 200)
The size of the region
used for motif finding is important. If analyzing
ChIP-Seq peaks from a transcription factor, Chuck would
recommend 50 bp for establishing the primary motif bound
by a given transcription factor and 200 bp for finding
both primary and "co-enriched" motifs for a
transcription factor. When looking at histone
marked regions, 500-1000 bp is probably a good idea
(i.e. H3K4me or H3/H4 acetylated regions). In
theory, HOMER can work with very large regions (i.e.
10kb), but with the larger the regions comes more
sequence and longer execution time. These regions
will be based off the center of the peaks. If you
prefer an offset, you can specify "-size -300,100" to
search a region of size 400 that is centered 100 bp
upstream of the peak center (useful if doing motif
finding on putative TSS regions). If you have
variable length regions, use the option "-size given"
and HOMER will use the exact regions that were used as
input.
Motif length ("-len <#>" or "-len <#>,<#>,...", default 8,10,12)
Specifies the length of
motifs to be found. HOMER will find motifs of each
size separately and then combine the results at the
end. The length of time it takes to find motifs
increases greatly with increasing size. In
general, it's best to try out enrichment with shorter
lengths (i.e. less than 15) before trying longer
lengths. Much longer motifs can be found with
HOMER, but it's best to use smaller sets of sequence
when trying to find long motifs (i.e. use "-len 20 -size
50"), otherwise it may take way too long (or take too
much memory). The other trick to reduce the total
resource consumption is to reduce the number of
background sequences (-N <#>).
Mismatches allowed in global optimization phase ("-mis <#>", default: 2)
HOMER looks for promising
candidates by initially checking ordinary oligos for
enrichment, allowing mismatches. The more
mismatches you allow, the more sensitive the algorithm,
particularly for longer motifs. However, this also
slows down the algorithm a bit. If searching for
motifs longer than 12-15 bp, it's best to increase this
value to at least 3 or even 4.
Number of CPUs to use ("-p <#>", default 1)
HOMER is now multicore
compliant. It's not perfectly parallelized,
however, certain types of analysis can benefit. In
general, the longer the length of the motif, the better
the speed-up you'll see.
Number of motifs to find ("-S <#>", default 25)
Specifies the number of
motifs of each length to find. 25 is already quite
a bit. If anything, I'd recommend reducing this
number, particularly for long motifs to reduce the total
execution time.
Normalize CpG% content instead of GC% content ("-cpg")
Consider tying if HOMER is
stuck finding "CGCGCGCG"-like motifs. You can also
play around with disabling GC/CpG normalization ("-noweight").
Region level autonormalization ("-nlen <#>", default 3, "-nlen 0" to disable)
Motif level autonormalization (-olen <#>, default 0 i.e. disabled)
Autonormalization attempts
to remove sequence bias from lower order oligos (1-mers,
2-mers ... up to <#>). Region level
autonormalization, which is for 1/2/3 mers by default,
attempts to normalize background regions by adjusting
their weights. If this isn't getting the job done
(autonormalization is not guaranteed to remove all
sequence bias), you can try the more aggressive motif
level autonormalization (-olen <#>). This performs
the autonormalization routine on the oligo table during
de novo motif discovery. (see here for more info)
User defined background regions ("-bg <peak file of background regions>")
Why let HOMER randomly
pick you background regions when you can choose them
yourself!! These will still be normalized for CpG% or
GC% content just like randomly chosen sequences and
autonormalized unless these options are turned off (i.e.
"-nlen 0 -noweight"). This can be very useful
since HOMER is a differential motif discovery
algorithm. For example, you can give HOMER a set
of peaks co-bound by another factor and compare them to
the rest of the peaks. HOMER will automatically
check if the background peaks overlap with the target
peaks using mergePeaks,
and discard overlapping regions.
Hypergeometric enrichment scoring ("-h")
By default, findMotifsGenome.pl
uses the binomial distribution to score motifs.
This works well when the number of background sequences
greatly out number the target sequences - however, if
you are using "-bg"
option above, and the number of background sequences is
smaller than target sequences, it is a good idea to use
the hypergeometric distribution instead ("-h").
FYI
-
The
binomial
is
faster to compute, hence it's use for motif finding in
large numbers of regions.
Find enrichment of individual oligos ("-oligo").
This creates output files
in the output directory named oligo.length.txt.
Force findMotifsGenome.pl to re-preparse genome for the given region size ("-preparse").
In case there is a problem
with the existing preparsed files, force them to be
remade with "-preparse".
Only search for motifs on + strand ("-norevopp")
By default, HOMER looks
for transcription factor-like motifs on both
strands. This will force it to only look at the +
strand (relative to the peak, so - strand if the peak is
on the - strand).
Search for RNA motifs ("-rna")
If looking at RNA data
(i.e. Clip-Seq or similar), this option will restrict
HOMER to only search the + strand (relative to the
peak), and will output RNA motif logos (i.e. U instead
of T). It will also try to compare found motifs to
an RNA motif database, which sadly, only contains miRNAs
right now... I guess chuck roundhouse kicked all of the
splicing and other RNA motifs into hard to find
databases.
Mask motifs ("-mask <motif file>")
Mask the motif(s) in the
supplied motif file before starting motif finding.
Multiple motifs can be in the motif file.
Optimize motifs ("-opt <motif file>")
Instead of looking for
novel de novo motifs, HOMER will instead try to optimize
the motif supplied. This is cool when trying to
change the length of a motif, or find a very long
version of a given motif. For example, if you
specify "-opt <file>" and "-len 50", it will try
to expand the motif to 50bp and optimize it.
Dump FASTA files ("-dumpFasta")
Like the fact that HOMER
organizes and extracts your sequence files, but don't
care for HOMER as a motif finding algorithm?
That's cool, just specify "-dumpFasta" and the files
"target.fa" and "background.fa" will show up in your
output directory. You can then use them with MEME
or whatever. Just remember, Chuck knows where you
live...
Finding Instance of Specific Motifs
By default, HOMER does not
return the locations of each motif found in the motif
discovery process. To recover the motif locations,
you must first select the motifs you're interested in by
getting the "motif file" output by HOMER. You can
combine multiple motifs in single file if you like to form
a "motif library". To identify motif locations, you
have two options:
1. Run findMotifsGenome.pl
with the "-find
<motif file>" option. This will
output a tab-delimited text file with each line
containing an instance of the motif in the target
peaks. The output is sent to stdout.
For example: findMotifsGenome.pl
ERalpha.peaks hg18 MotifOutputDirectory/ -find
motif1.motif > outputfile.txt
The output file will contain the columns:
2. Run annotatePeaks.pl
with the "-m <motif
file>" option (see the annotation section for more
info). Chuck prefers doing it this way. This
will output a tab-delimited text file with each line
containing a peak/region and a column containing
instance of each motif separated by commas to stdoutThe output file will contain the columns:
- Peak/Region ID
- Offset from the center of the region
- Sequence of the site
- Name of the Motif
- Strand
- Motif Score (log odds score of the motif matrix,
higher scores are better matches)
For example: annotatePeaks.pl
ERalpha.peaks hg18 -m motif1.motif >
outputfile.txt
The output file will contain columns:
19. CpG%
20. GC%
21. Motif Instances
...
Motif Instances have the following format:
The output file will contain columns:
- Peak/Region ID
- Chromosome
- Start
- End
- Strand of Peaks
19. CpG%
20. GC%
21. Motif Instances
...
Motif Instances have the following format:
<distance from
center of
region>(<sequence>,<strand>,<conservation>)
i.e -29(TAAATCAACA,+,0.00)
i.e -29(TAAATCAACA,+,0.00)
You can also find histogram
of motif density this way by adding "-hist <#>" to
the command. For example:
Graphing the output with EXCEL:
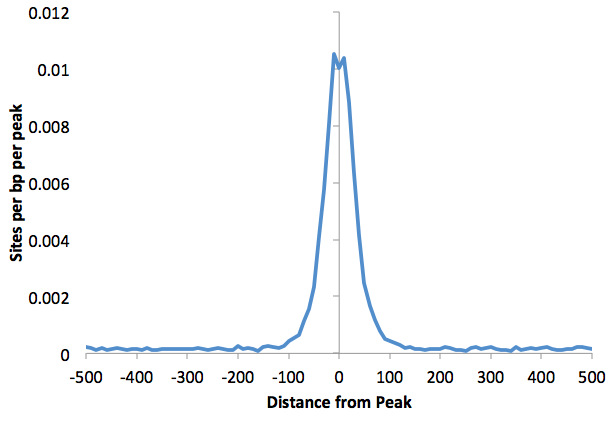
annotatePeaks.pl ERalpha.peaks hg18 -m
ere.motif foxa1.motif -size 1000 -hist 10
> outputfile.txt
Graphing the output with EXCEL:
Command-line options for findMotifsGenome.pl
Program will find de novo and known motifs in regions in the genomeUsage: findMotifsGenome.pl <pos file> <genome> <output directory> [additional options]
Example: findMotifsGenome.pl peaks.txt mm8r peakAnalysis -size 200 -len 8
Possible Genomes:
...
Custom: provide the path to genome FASTA files (directory or single file)
Heads up: will create the directory "preparsed/" in same location.
Basic options:
-bg <background position file> (genomic positions to be used as background, default=automatic)
removes background positions overlapping with target positions
-chopify (chop up large background regions to the avg size of target regions)
-len <#>[,<#>,<#>...] (motif length, default=8,10,12) [NOTE: values greater 12 may cause the program
to run out of memory - in these cases decrease the number of sequences analyzed (-N),
or try analyzing shorter sequence regions (i.e. -size 100)]
-size <#> (fragment size to use for motif finding, default=200)
-size <#,#> (i.e. -size -100,50 will get sequences from -100 to +50 relative from center)
-size given (uses the exact regions you give it)
-S <#> (Number of motifs to optimize, default: 25)
-mis <#> (global optimization: searches for strings with # mismatches, default: 2)
-norevopp (don't search reverse strand for motifs)
-nomotif (don't search for de novo motif enrichment)
-rna (output RNA motif logos and compare to RNA motif database, automatically sets -norevopp)
Scanning sequence for motifs
-find <motif file> (This will cause the program to only scan for motifs)
Known Motif Options/Visualization
-bits (scale sequence logos by information content, default: doesn't scale)
-nocheck (don't search for de novo vs. known motif similarity)
-mcheck <motif file> (known motifs to check against de novo motifs,
default: /bioinformatics/homer/data/knownTFs/all.motifs
-float (allow adjustment of the degeneracy threshold for known motifs to improve p-value[dangerous])
-noknown (don't search for known motif enrichment, default: -known)
-mknown <motif file> (known motifs to check for enrichment,
default: /bioinformatics/homer/data/knownTFs/known.motifs
Sequence normalization options:
-gc (use GC% for sequence content normalization, now the default)
-cpg (use CpG% instead of GC% for sequence content normalization)
-noweight (no CG correction)
Advanced options:
-h (use hypergeometric for p-values, binomial is default)
-N <#> (Number of sequences to use for motif finding, default=max(50k, 2x input)
-noforce (will attempt to reuse sequence files etc. that are already in output directory)
-local <#> (use local background, # of equal size regions around peaks to use i.e. 2)
-redundant <#> (Remove redundant sequences matching greater than # percent, i.e. -redundant 0.5)
-mask <motif file1> [motif file 2]... (motifs to mask before motif finding)
-opt <motif file1> [motif file 2]... (motifs to optimize or change length of)
-refine <motif file1> (motif to optimize)
-rand (randomize target and background sequences labels)
-ref <peak file> (use file for target and background - first argument is list of peak ids for targets)
-oligo (perform analysis of individual oligo enrichment)
-dumpFasta (Dump fasta files for target and background sequences for use with other programs)
-preparse (force new background files to be created)
-keepFiles (keep temporary files)
homer2 specific options:
-homer2 (use homer2 instead of original homer, default)
-nlen <#> (length of lower-order oligos to normalize in background, default: -nlen 3)
-nmax <#> (Max normalization iterations, default: 160)
-olen <#> (lower-order oligo normalization for oligo table, use if -nlen isn't working well)
-p <#> (Number of processors to use, default: 1)
-e <#> (Maximum expected motif instance per bp in random sequence, default: 0.01)
-cache <#> (size in MB for statistics cache, default: 500)
-quickMask (skip full masking after finding motifs, similar to original homer)
Original homer specific options:
-homer1 (to force the use of the original homer)
-depth [low|med|high|allnight] (time spent on local optimization default: med)

Can't figure something out? Questions, comments, concerns, or other feedback:
cbenner@salk.edu