
HOMER
Software for motif discovery and ChIP-Seq analysis
Next-gen Sequencing Analysis: Creating a "Tag Directory"
with makeTagDirectory
To facilitate the analysis of ChIP-Seq (or any other type of
short read re-sequencing data), it is useful to first
transform the sequence alignment into platform independent
data structure representing the experiment, analogous to
loading the data into a database. HOMER does this by
placing all relevant information about the experiment into a
"Tag Directory", which is essentially a directory on your
computer that contains several files describing your
experiment. In many ways tag directories are
like sorted bam files, and some day soon Chuck promises
HOMER will work directly with sorted bam files too...
(Unfortunately, at present bam files must be converted into
tag directories).During the creation of tag directories, several quality control routines are run to help provide information and feedback about the quality of the experiment. During this phase several important parameters are estimated that are later used for downstream analysis, such as the estimated length of ChIP-Seq fragments.
Input Alignment Files
To create a "Tag Directory", you must have alignment files in one of the following formats:
- BED format
- SAM format
- BAM format (HOMER will use "samtools view file.BAM
> file.SAM" to covert to a SAM file, so "samtools"
must be available)
- default bowtie output format
- *.eland_result.txt or *_export.txt format from the Illumina pipeline
Column2: start position
Column3: end position
Column4: Name (or strand +/-)
Column5: Ignored**
Column6: Strand +/-
**Unfortunately, BED files are used in different ways by different groups. By default, HOMER ignores the 5th column of BED files. In some cases the 5th column is used to encode the number of reads at that position. If this is the case, use the "-force5th".
Alternatively (or in combination), you can make tag directories from existing tag directories or from tag files (explained below).
If your input files are named "s_1_sequence.txt", or have a suffix such as *.fq or *.fastq, then you probably have raw sequence files without sequence alignment information. You need to align these sequences to the genome first. Learn about aligning data to the genome here.
Specialized Input Files
As HOMER's capabilities expand, more and more file formats are being added. Below is the current list:
- DNA methylation data: Files for mCpG data (coming soon).
- Hi-C summary format: Find out more about this in the Hi-C section.
Paired-End Reads
Creating Tag Directories
An example:
Several additional options exist for makeTagDirectory. The program attempts to guess the format of your alignment files, but if it is unsuccessful, you can force the format with "-format <X>".
makeTagDirectory Macrophage-H3K4me1-ChIP-Seq/ s_1_sequence.align.sam -format sam
Sometimes BED file alignments contain stupid values in the 5th column, such as quality information etc. HOMER will treat this value as the number of reads aligning to the same location. If this is not how the value is used, add "-forceBED" to ignore the value found int the 5th column of a BED file.
To combine tag directories, for example when combining two separate experiments into one, do the following:
Filtering Reads/Selecting Uniquely Alignable Reads from SAM/BAM files
The makeTagDirectory program has several specialized options for processing SAM/BAM formatted files. A quick introduction to SAM/BAM files can be found here. Because of the various ways aligner create SAM/BAM files, it is often easiest to have HOMER filter the reads that are not uniquely alignable, etc. Below is a list of the various options that are available to process SAM/BAM files:
-unique : selecting this option will cause HOMER to only keep uniquely alignable reads (this is also the default). For HOMER to consider a read unique, it must NOT have the "secondary alignment" flag set, and it's MAPQ score must be greater than 10. To change the MAPQ cutoff, use the "-mapq <#>" option. If you set the option to "-mapq -1", HOMER will try to read AS:i:# and XS:i:# flags (if set) to figure out if the secondary alignment score is the same or less than the alignment.There are also ways to filter/select reads with specific properties:
-keepOne : selecting this will cause HOMER to keep all primary alignments, regardless of MAPQ score. Alignments with the "secondary alignment" flag set will be discarded.
-keepAll : this will cause HOMER to keep all alignment in the SAM/BAM file.
-mis <#> : Maximum allowable mismatches allowed in the read. Only works if the MD:Z: flag is set.
-tbp <#> : only count one read per position per length of the read (i.e. if one read is 100 bp long, an another 75 bp long, but start at the same position, both with be kept).
-minlen <#> : minimum length of the read to keep (useful for small RNA processing, i.e. keeping 22-23 bp sequeces for miRNAs, 26-32 bp for piRNA, etc.)
-maxlen <#> : maximum length of read to keep (use in conjunction with -minlen <#>)
-filterReads <seq> <offset> <keep | remove> : Advanced read filtering based on the presence of a specific oligo sequence in the genome (REQUIRES "-genome <genome>"). For example:
For example: -filterReads GATC -4 keep
This will only keep reads that contain a GATC sequence 4 bp upstream from the start of the read.
Interpreting the correct strand
By default, HOMER will interpret each read as it is presented in the alignment file. This can differ from the expected results if you have paired-end sequencing from a strand-specific RNA-Seq experiment. It can also result in reads aligning to the opposite strand if a 'first-strand' library synthesis protocol is used, such as methods that use the deoxy-UTP incorporation. To control how HOMER interprets the strands you have two choices: Either use "-strand -" when running programs like annotatePeaks.pl when you would normally use "-strand +", or you can flip the strand during the makeTagDirectory step:
-flip : (will flip the strands of each read in the alignment file)For example, if you're analyzing data from Illumina's newer strand specific RNA-Seq kits, you'll probably want to use "-flip" and also "-sspe" if you have a paired-end experiment.
-sspe : (for Strand Specific Paired End sequencing - this will flip the 2nd read of the mate-pair to match the first read).
What does makeTagDirectory do?
In the end, your output directory will contain several *.tags.tsv files, as well as a file named "tagInfo.txt". This file contains information about your sequencing run, including the total number of tags considered. This file is used by later programs to quickly reference information about the experiment, and can be manually modified to set certain parameters for analysis.
Additional options to control the behavior of makeTagDirectory:
-tbp <#> : Limit the number of tags per base pair to this number (default: no limit). For example, if you want to remove all duplicate reads that start at the same positions, specify "-tbp 1".makeTagDirectory also performs several quality control steps shown below.
-precision <1|2|3> : Set the number of decimal places used to store read information. The default is 1, which keeps the file size down. If storing methyl-cytosine information, it's better to increase the precision (this is automated when possible)
-single : Setting this option will place all reads into a single tag file instead of separate tag files for each chromosome. This is necessary for genome assemblies composed of thousands of scaffolds. HOMER will usually inform you if it wants you to use this option.
-totalReads <# | all | default> : The total reads in the tag directory is important for downstream normalization. This option allows you to modify this value for specific needs. Normally, only the reads that uniquely align to the genome are counted toward the total reads. However, you may want to include the total reads (unique+multimappers) without having the multimappers in the tag directory. You can do this by specifying "-totalReads all". You can also specify the total directly by providing a number.
Basic Quality Control Analysis:
makeTagDirectory ExistingTagDirectory/ -update [additional options...]
Basic Tag information
Read Length Distribution
Clonal Tag Distribution
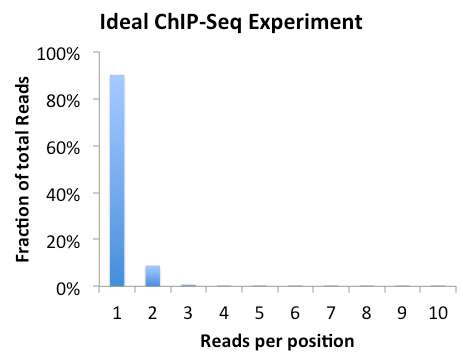
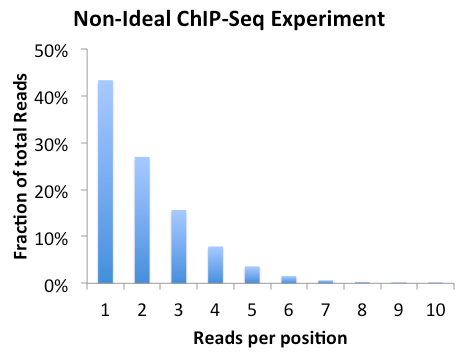
If the experiment is highly clonal and not expected to be, it might help to clean up the downstream analysis by forcing tag counts at each position to be no greater than x, where x is usually 1. To do this rerun the makeTagDirectory command and add "-tbp <#>" where # is the maxium tags per bp.
Autocorrelation Analysis
Different types of experiments (i.e. ChIP-Seq vs. DNase-Seq) produce different looking autocorrelation plots, and more detailed discussion of these differences can be found in the individual tutorials. Below is an example from a successful ChIP-Seq experiment:
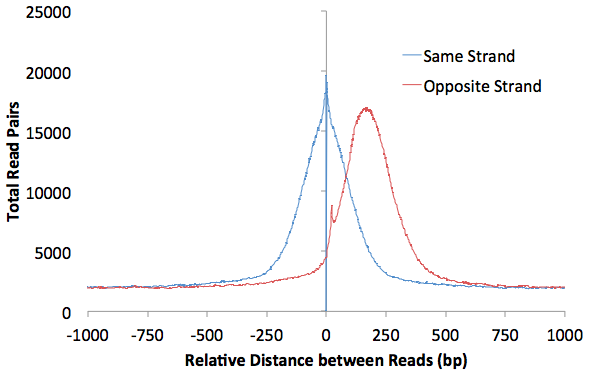
HOMER also uses the autocorrelation results to guess what type of experiment you conducted. It computes 3 statistics:
- Same strand fold enrichment: Enrichment of reads on the same strand within 3x the estimated fragment length
- Diff strand fold enrichment: Enrichment of reads on different strands within 3x the estimated fragment length
- Same / Diff fold enrichment: Difference between enrichment of reads on the same strand or different strands
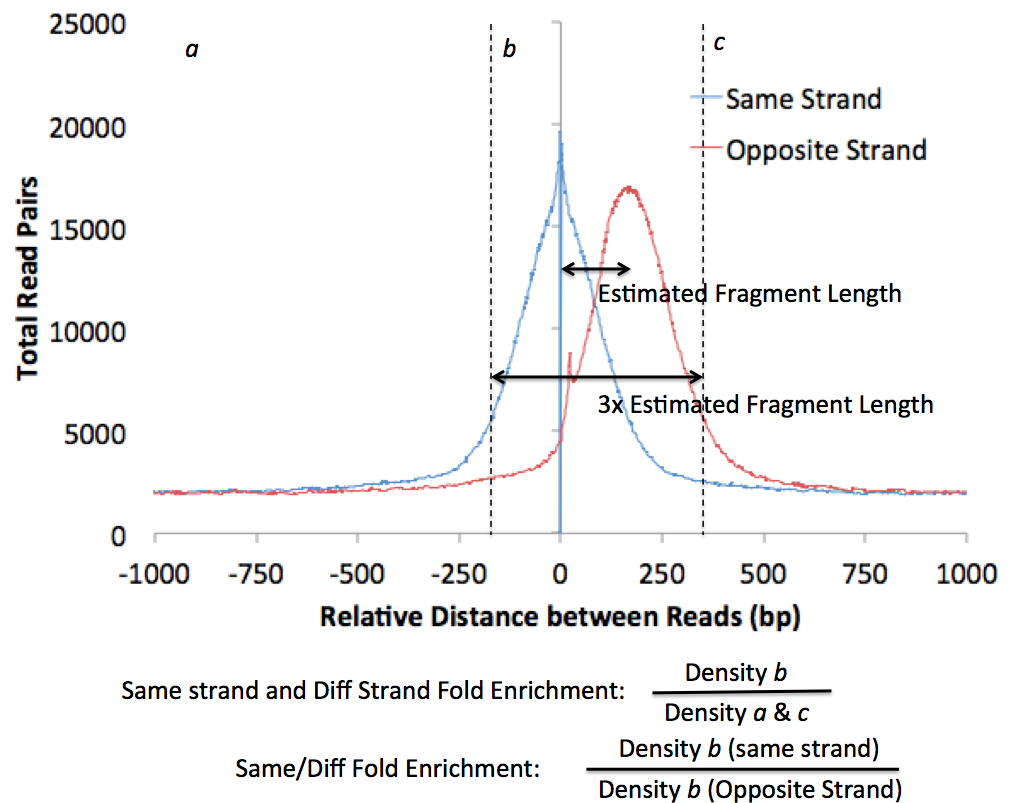
- If the Same/Diff Fold Enrichment is > 8 fold,
it's a great chance the sample is strand-specific
RNA. If HOMER decides the sample is probably
RNA due to the difference between same strand and
different strand numbers, it will automatically set
the estimated fragment length to 75 bp. This
is because it is difficult to estimate the fragment
length for RNA/GRO-seq. To manually set the
fragment length, use "-fragLength <#>"
- If both the "Same Strand Fold Enrichment" and "Diff Strand Fold Enrichment" are both greater than 1.5 fold, there is good chance you're looking at a working ChIP-Seq experiment.
Sequence Bias Analysis:
i.e. makeTagDirectory Macrophage-PU.1-ChIP-Seq -genome mm9 -checkGC pu1.alignment.bed
This analysis will produce several output files in addition to the basic quality control analysis described above.
An important prerequisite for analyzing sequence bias is that the appropriate genome must by configured for use with HOMER. In version v3.1, HOMER now handles custom/arbitrary genomes. Instead of intalling/configuring a genome, you can specify the path to a file or directory containing the genomic sequence in FASTA format. The genome can be in a single FASTA file, or you specify a directory where where each chromosome is in a separate file (named chrXXX.fa or chrXXX.fa.masked). In either case, the FASTA headers must contain the chromosome names followed by white space, i.e. ">chr blahblahblah", not ">chr1-blahblahblah", or prefereably only ">chr1".
NOTE: If using a sequencing type other than ChIP-Seq or MNase-Seq, you may want to add "-fragLength <#>" since it may be difficult for HOMER to automatically determine the size of fragments used for sequencing.
Genomic Nucleotide Frequency relative to read
positions
tagFreqUniq.txt - Same as tagFreq.txt, however individual genomic positions are only counted once. If 10 reads mapped to the same position, those nucleotide counts would be added 10 times for "tagFreq.txt", but only once for "tagFreqUniq.txt".
By default, HOMER calculates this frequency from -50 bp to +50 bp relative to the end of the estimated fragment (e.g. not +50bp relative to the 36 bp read, but +50 bp relative to the 200 bp ChIP-fragment). This can be changed using the options "-freqStart <#>" and "-freqEnd <#>"
Below are some examples of this file graphed with EXCEL. One is a pretty typical ChIP-Seq file, the other Chuck wouldn't say where it came from, but he's pretty sure there might be a problem with how they performed their experiment... Quite a bit can be learned from looking at these types of plots (see individual tutorials of additional details)
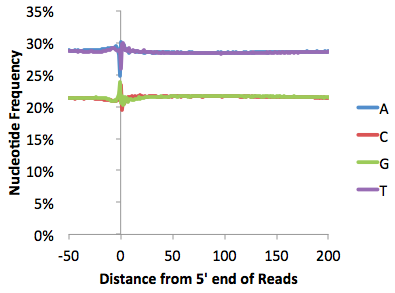
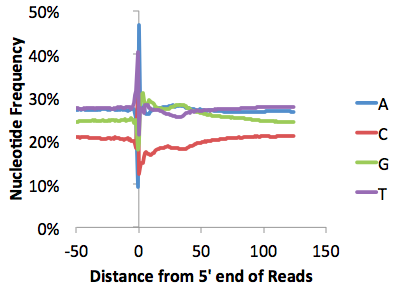
Fragment GC% Distribution
genomeGCcontent.txt - Distribution of fragment GC%-content at each location in the genome (i.e. expected distribution)
These are very important files to check for any sequencing experiment. Due to the numerous steps of library preparation involving amplification, size selection and gel extraction, it is very easy for the average GC%-content of the sequencing library to "shift". This can be a disastrous problem. Consider the following:
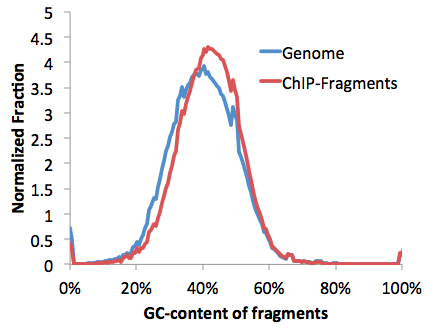
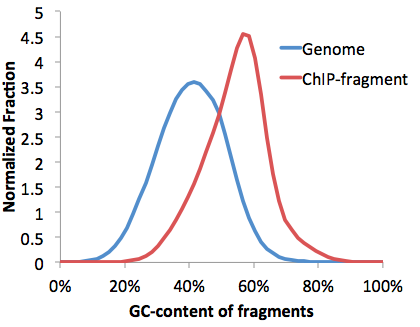
The problem with a GC% shifted sample is that even if the sample is random sequence, you will start to show "enrichment" at places with high GC-content in the genome, such as at CpG Islands. This is unfortunate because most GC-rich areas are at transcription start sites, which might make you think the experiment worked, when in reality the sample was boiled instead of placed in the freezer. See below to learn about GC-normalization.
Sequence Bias GC normalization:
When should I normalize for GC-content in my sequencing experiment?
For some experiments, it tough to know what the expected GC-content should be. For example, if sequencing H3K4me3 ChIP-Seq data, which is typically found near CpG Islands, you may expect the GC% to be higher. However, even with H3K4me3 ChIP-Seq, most ChIP experiments are not very efficient, and most of the DNA being sequenced is background. As a result, the average GC content should not be too far off the expected genomic average. You can always "try" normalizing.
If the GC-bias is severe, you might be better off repeating the experiment. Normalization essentially destroys information, so repeating the experiment (or at least the sequencing library preparation) may be a good move. When re-preping the sample, try amplifying less and when extracting DNA from gels, dissolve the gels at room temperature.
Normalizing tags for GC-bias
i.e. makeTagDirectory Macrophage-PU.1-GC -genome mm9 -normGC default -iterNorm 0.01 aligned.reads.bed
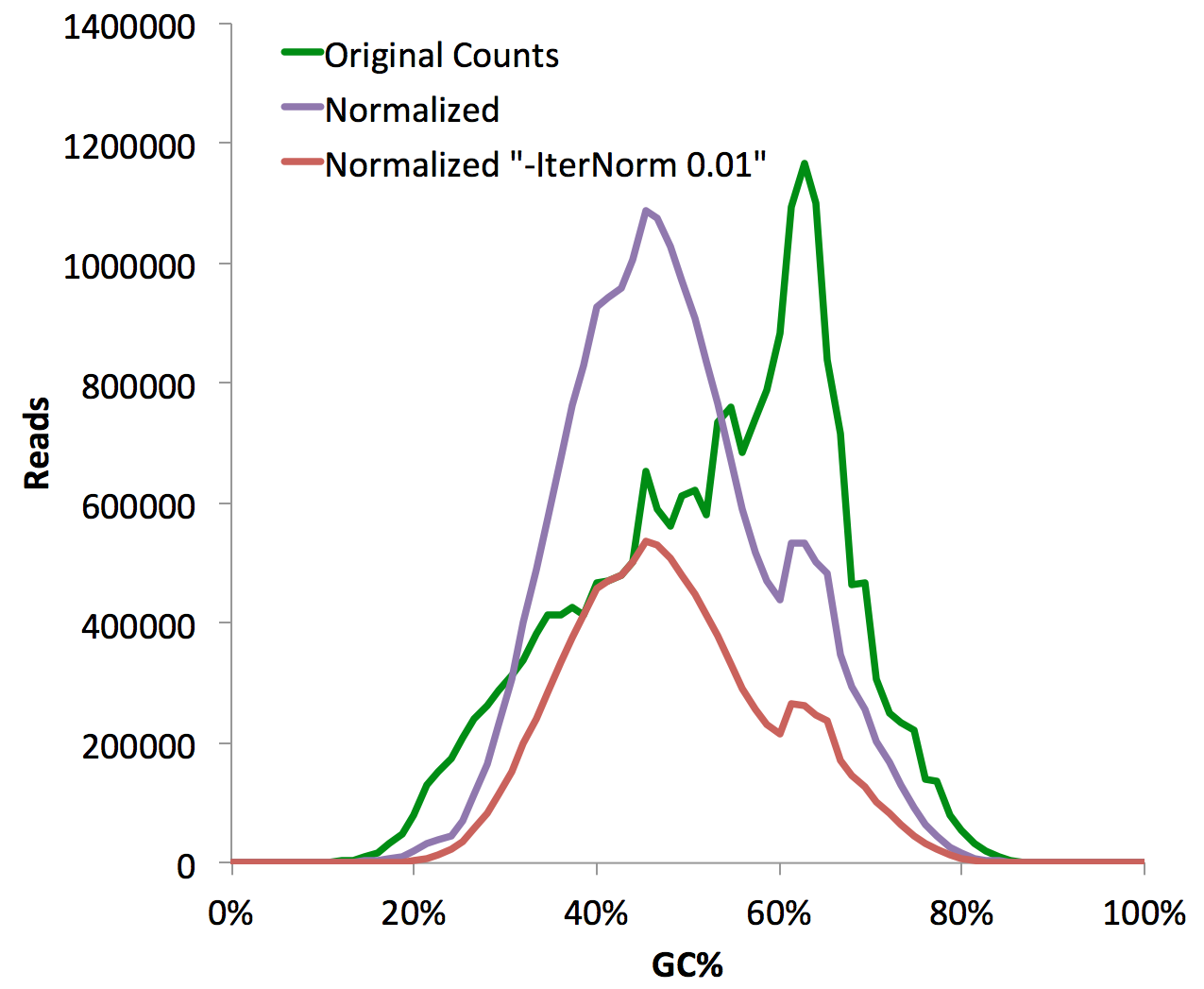
The new GC normalization procedure attempts to be conservative by only normalizing by 'removing' reads from the experiment by down weighting the read counts associated with each position. It is dangerous to mathematically "add reads" to the experiment - this is sort of like creating information which we don't really have, particularly for later if you are doing count-based differential expression. HOMER will remove iteratively remove reads from GC ranges that are overrepresented until the overall distribution of GC is with the indicated percentage of the "-iterNorm <#>" parameter. Of course, it doesn't actually remove the reads, just reduces them to fractional values.
The alternative/original GC normalization procedure is actually very simple. HOMER calculates the distribution of fragment GC%-contents, then for each range of GC%, normalizes the tag counts to the expected distribution. If your sample is more GC-rich that expected, reads in GC-rich regions will be reduced to a fractional value (limited by "-minNormRatio <#>"). Likewise, if reads are in AT-rich regions, their values will be increased (limited by "-maxNormRatio <#>"). HOMER is happy to deal with fractional tag values - there is no reason why a sequencing read can't be counted as a "half-a-read", for example. The biggest problem problem with increasing the value of a read is that it is akin to creating information that doesn't exist, so HOMER puts a tight cap on that adjustment to only 2-fold (change with "-maxNormRatio <#>"). The resulting normalization information is recorded in the tagGCnormalization.txt file. To enact this form of GC normalization, omit the "-iterNorm <#>" parameter.
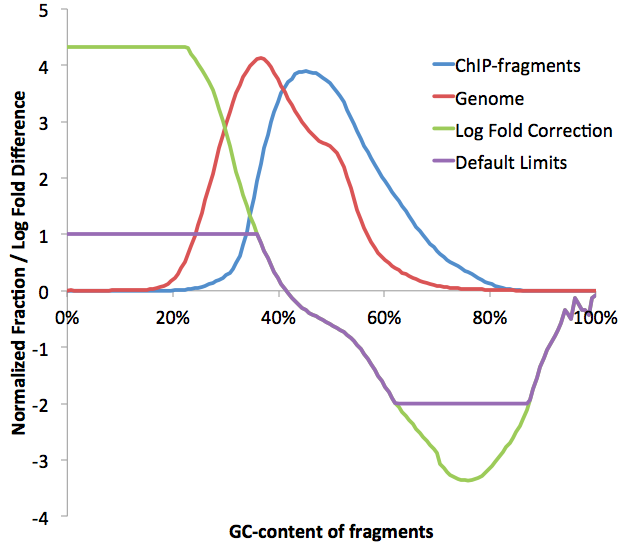
Often, the expected genome GC-content is not appropriate. To normalize to a custom GC-profile, provide a file after -normGC option ("-normGC <filename>"). This file should be formatted just like the tagGCcontent.txt files found in the directories. In fact, the idea is that you can normalize one experiment to another by providing the experiments tagGCcontent.txt file as the argument for -normGC. One potential strategy is to combine several experimental files across different experiments into a single tag directory, and then use the tagGCcontent.txt file from this directory to normalize each of the other experiment, normalizing each experiment to the average from all of the experiments.
The difference between the two normalization strategies can be seen in the resulting read counts of the experiment (the -iterNorm <#> method essentially removes reads until it can match the distribution):
Command line options of makeTagDirectory command:
To perform QC/manipulations on an existing tag directory, add "-update"Options:
-fragLength <# | given> (Set estimated fragment length - given: use read lengths)
By default treats the sample as a single read ChIP-Seq experiment
-format <X> where X can be: (with column specifications underneath)
bed - BED format files:
(1:chr,2:start,3:end,4:+/- or read name,5:# tags,6:+/-)
-force5th (5th column of BED file contains # of reads mapping to position)
sam - SAM formatted files (use samTools to covert BAMs into SAM if you have BAM)
-unique (keep if there is a single best alignment based on mapq)
-mapq <#> (Minimum mapq for -unique, default: 10, set negative to use AS:i:/XS:i:)
-keepOne (keep one of the best alignments even if others exist)
-keepAll (include all alignments in SAM file)
-mis (Maximum allowed mismatches, default: no limit, uses MD:Z: tag)
-sspe (strand specific, paired-end reads[flips strand of 2nd read to match])
bowtie - output from bowtie (run with --best -k 2 options)
(1:read name,2:+/-,3:chr,4:position,5:seq,6:quality,7:NA,8:misInfo)
eland_result - output from basic eland
(1:read name,2:seq,3:code,4:#zeroMM,5:#oneMM,6:#twoMM,7:chr,
8:position,9:F/R,10-:mismatches
eland_export - output from illumina pipeline (22 columns total)
(1-5:read name info,9:sequence,10:quality,11:chr,13:position,14:strand)
eland_extended - output from illumina pipeline (4 columns total)
(1:read name,2:sequence,3:match stats,4:positions[,])
mCpGbed - encode style mCpG reporting in extended BED format, no auto-detect
(1:chr,2:start,3:end,4:name,5:,6:+/-,7:,8:,9:,10:#C,11:#mC)
allC - Lister style output files detailing the read information about all cytosines
(1:chr,2:pos,3:strand,4:context,#mC,#totalC,#unmC
bismark - Bismark style output files detailing the read information about all cytosines
(1:chr,2:pos,3:strand,4:#mC,5:#unmC,6:context,7:triseq
-minCounts <#> (minimum number of reads to report mC/C ratios, default: 10)
-mCcontext <CG|CHG|CHH|all> (only use C's in this context, default: CG)
HiCsummary - minimal paired-end read mapping information
(1:readname,2:chr1,3:5'pos1,4:strand1,5:chr2,6:5'pos2,7:strand2)
-flip (flip strand of each read, i.e. might want to use with some RNA-seq)
-totalReads <#|all|default> (set the effective total number of reads - all includes multimappers)
-force5th (5th column of BED file contains # of reads mapping to position)
-d <tag directory> [tag directory 2] ... (add Tag directory to new tag directory)
-t <tag file> [tag file 2] ... (add tag file i.e. *.tags.tsv to new tag directory)
-single (Create a single tags.tsv file for all "chromosomes" - i.e. if >100 chromosomes)
-update (Use current tag directory for QC/processing, do not parse new alignment files)
-tbp <#> (Maximum tags per bp, default: no maximum)
-precision <1|2|3> (number of decimal places to use for tag totals, default: 1)
-minlen <#> and -maxlen <#> (Filter reads with lengths outside this range)
GC-bias options:
-genome <genome version> (To see available genomes, use "-genome list")
-or- (for custom genomes):
-genome <path-to-FASTA file or directory of FASTA files>
-checkGC (check Sequence bias, requires "-genome")
-freqStart <#> (offset to start calculating frequency, default: -50)
-freqEnd <#> (distance past fragment length to calculate frequency, default: +50)
-oligoStart <#> (oligo bias start)
-oligoEnd <#> (oligo bias end)
-normGC <target GC profile file> (i.e. tagGCcontent.txt file from control experiment)
Use "-normGC default" to match the genomic GC distribution
-normFixedOligo <oligoFreqFile> (normalize 5' end bias, "-normFixedOligo default" ok)
-minNormRatio <#> (Minimum deflation ratio of tag counts, default: 0.25)
-maxNormRatio <#> (Maximum inflation ratio of tag counts, default: 2.0)
-iterNorm <#> (Sets -max/minNormRatio to 1 and 0, iteratively normalizes such that the
resulting distrubtion is no more than #% different than target, i.e. 0.1,default: off)
-filterReads <seq> <offset> <keep|remove> (filter reads based on oligo sequence in the genome)
HiC options:
-bowtiePE (when matching PE reads in bowtie alignment format, assumes last character of read name is 0 or 1)
-removePEbg (remove paired end tags within 1.5x fragment length on same chr)
-PEbgLength <#> (remove PE reads facing on another within this distance, default: 1.5x fragLen)
-restrictionSite <seq> (i.e. AAGCTT for HindIII, assign data < 1.5x fragment length to sites)
Must specify genome sequence directory too. (-rsmis <#> to specify mismatches, def: 0)
-both, -one, -onlyOne, -none (Keeps reads near restriction sites, default: keep all)
-removeSelfLigation (removes reads linking same restriction fragment)
-removeRestrictionEnds (removes reads starting on a restriction fragment)
-assignMidPoint (will place reads in the middle of HindIII fragments)
-restrictionSiteLength <#> (maximum distance from restriction site, default: 1.5x fragLen)
-removeSpikes <size bp> <#> (remove tags from regions with > than # times
the average tags per size bp, suggest "-removeSpikes 10000 8")
Next: Creating UCSC Genome Browser
visualization files

Can't figure something out? Questions, comments, concerns, or other feedback:
cbenner@salk.edu