
HOMER
Software for motif discovery and next-gen sequencing analysis
Visualizing Experiments with a Genome Browser
The
UCSC Genome Browser is
quite possibly one of the best computational tools ever
developed. Not only does it contain an incredible
amount of data in a single application, it allows users to
upload custom information such as data from their ChIP-Seq
experiments so that they can be easily visualized and
compared to other information. There are also other
genome browsers that are available, and each has a different
strength:UCSC Genome Browser
Truly a unique resource, logs of data preloaded and annotations.WashU Epigenome Browser
Capable of visualizing long-range interactions (great for data sets like Hi-C), also has a lot of preloaded data.IGV
The Integrated Genomics Viewer (IGV), great for looking at reads locally instead of needing to load them to a server/cloud based solution. Great for directly looking at sorted bam/bai files to examine mutations in reads.Many others...
Most of the tools that are part of HOMER cater to the
strengths of the UCSC Genome Browser - however, the
bedGraph and other files generated by HOMER can be
normally be used in the other genome browsers as well.
Making Genome Browser Files
i.e. makeUCSCfile PU.1-ChIP-Seq/ -o auto
(output file will be in the PU.1-ChIP-Seq/ folder named PU.1-ChIP-Seq.ucsc.bedGraph.gz)
To visualize the experiment in the UCSC Genome Browser, go to Genome Browser page and select the appropriate genome (i.e. the genome that the sequencing tags were mapped to). Then click on the "add custom tracks" button (this will read "manage custom tracks" once at least one custom track is loaded). Enter the file created earlier in the "Paste URLs or data" section and click "Submit".
Problems Loading UCSC Files
2. Did you align the genome to a UCSC version? chr1 != Chr1 != 1
3. Some of your tags are mapping outside the reference chromosome - this can be caused by mapping to non-standard assemblies or by some alignment programs. To remove all reads outside of the UCSC chromosome lengths, you can run the program removeOutOfBoundsReads.pl.
i.e. removeOutOfBoundsReads.pl PU.1-ChIP-Seq/ mm9
After running the program, you can rerun makeUCSCfile.
What does makeUCSCfile do?
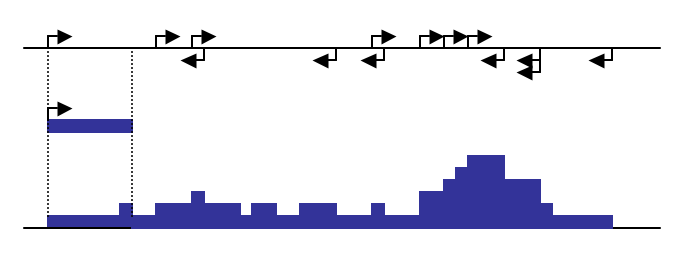
As great as the UCSC Genome Browser is, the large size of recent ChIP-Seq experiments results in custom track files that are very large. In addition to taking a long time to upload, the genome browser has trouble loading excessively large files. To help cope with this, the makeUCSCfile program can help by specifying a target file size when zipped (i.e. "-fsize 50e6" for 50MB). In order to meet the specified target file size, makeUCSCfile merges adjacent regions of tag density levels by their weighted average to reduce the total number lines in the final bedGraph file. If you have trouble loading getting your file to load, try reducing the size of the file using the "-fsize <#>" option (i.e. "-fsize 2e7"). To force the creation of larger files, use a very large file size (i.e. "-fsize 1e50") - this will create a file that does not merge any regions and displays a "native" view of the data.
Tags can be visualized separately for each strand using the "-strand separate" option.
Changing the Resolution
In an effort to reduce the size of large UCSC files, one attractive option is to reduce the overall resolution of the file. By default, makeUCSCfile will make full resolution (i.e. 1 bp) files, but this can be changed by specifying the "-res <#>" option. For example, "-res 10" will cause changes in ChIP-fragment density to be reported only every 10 bp.
Normalization of UCSC files: 2 types of normalization
-norm <#> : Normalize the total number of reads to this number, default 1e7. This means that tags from an experiment with only 5 million mapped tags will count for 2 tags apiece.
-normLength <#> : Set the standand length for normalization, default 100.
"-normLength 0" will disable the length normalization altogether, useful when visualizing single nucleotide data.-noadj or -raw : who needs normalization? Just give me the raw coverage numbers...
Normalizing files to Input
The paragraph above specifies how to normalize read densities based on the total number of reads. For some applications, particularly if studying organisms with small genomes, it is better to visualize the read density as a ratio relative to Input or IgG. Normally I would NOT recommend visualizing reads this way if the Input/IgG read coverage is sparse as this will cause trouble when calculating ratios. To normalize the experiment to a second tag directory, use the "-i <input tag directory>" option:
makeUCSCfile ExpTagDirectory/ -i InputTagDirectory -o autoAdditional parameters to control the normalized output:
-pseudo <#> : To avoid fluctuations in the ratio due to low coverage input, a pseudo count is added to the numerator and denominator when calculating the ratio (default: 5)
-log : report as a log ratio (default is a simple ratio)
-inputtbp <#> : set the maximum tags per bp considered in the input experiment
Separating data from different strands / RNA-Seq
-strand separate : separate data by strand, for RNA-Seq/GRO-Seq
-strand + : only show the positive strand (i.e. Watson strand) data
-strand - : only show the negative strand (i.e. crick strand) data
RNA-Seq and Splicing:
HOMER does not fully support the visualization of spliced RNA-Seq reads. However, if you specify the "-fragLength given" option, HOMER will only visualize the reads from the 5' end of the read until the first splice site (or the end of the read). This will help make the read densities look nice a crisp over exons, but will not visualize parts of the read that are 3' from the first splice found in the read.
Modifying Read Coverage
You can manually set the fragment lengths that are visualized and shift their positions, both of which can be useful:
-fragLength <# | given> : sets the fragment length, default: uses fragmentLengthEstimate in the tagInfo.txt file of the tag directory. If you want to visualize how the signal changes over large regions, it can be useful to set the fragment length to a very large value (i.e. 10000). If you want to visualize the exact length of the reads, use "-fragLength given".
-adjust <#> : adjust the position of the read by this amount from the 5' end. For example, -adjust -10 would start the coverage 10 bp upstream. This useful when the 5' end of the read represents a localized signal, i.e. DNase nicking site, as opposed to a ChIP-Seq fragment, which implies the factor binds downstream from the 5' end.
-tbp <#> : limit the number of reads considered per position, default: no limit. i.e. "-tbp 1" only counts one read per position.
-inputFragLength <#>, -inputAdjust <#>, -inputtbp <#> work the same for input directories if calculating a ratio.
Special Visualization Styles
To help streamline the visualization of different data types, you can use the "-style <styles>" option (i.e. "-style rnaseq"). This will adjust parameters for each type:
chipseq : standard, default
rnaseq : strand specific, will only extended fragments their given amount to help visualize exon edges.
tss : strand specific, and only shows the 5' nucleotide of the read (single base precision)
dnase : for nicking style DNase data only (see here), centers read fragment over the 5' end of the read.
methylated : reports cytosine methylation percentage at single bp resolution.
unmethylated : reports the percentage of unmethylated cytosinse at single bp resolution.
damid : reports large coverage fragments (2kb) centered on 5' end of the reads
Creating bigWig files with HOMER
Before even trying to make bigWigs, you must download the bedGraphToBigWig program from UCSC and place it somewhere in your executable path (i.e. the /path-to-homer/bin/ folder). This called directly by HOMER to create the BigWig files.
Using the makeBigWig.pl Script
i.e. makeBigWig.pl PU.1-ChIP-Seq/ mm9 -webDir /var/www/bigWigs/ -url http://ChuckNorrisU.edu/bigWigs/
If you are visualizing strand specific data (i.e. RNA-Seq), specify "-strand". The -url and -webDir are the directories are the web URL directory and file system directory where the bigWigs will be stored, respectively. Recent changes to UCSC require that the chromosome sizes be specified exactly. If having trouble, the current version of HOMER has the option "-chromSizes <filename>" so that you can specify the sizes explicitly.
Other makeBigWig.pl options:
-normal (default, similar to "-style chipseq" for makeUCSCfile).
-strand (for RNAseq, will create two bigWigs separately for each strand).
-dnase (will use "-style dnase")
-cage (combines -strand with -style cage)
-cpg (creates both methylated and unmethylated bigWigs)
-update (will overwrite default bigWig for that tag directory name. Otherwise, if the same file name exists, a random number will be added to the end)
-chromSizes <chrom.size file> (specify the chromosome sizes, default: automatic)
-url <URL> (URL directory -no filename- to tell UCSC where to look)
-webdir <directory> (name of directory to place resulting bigWig file)
Making bigWigs from scratch
After running the makeUCSCfile program with the bigWig options, you need to do the following:
- Copy the *.bigWig file to your webserver location and make sure it is viewable over the internet.
- Need to edit the "trackfileoutput.txt" file and enter the URL of your bigWig file (... bigDataUrl=http://server/path/bigWigFilename ...)
- Upload the "trackfileoutput.txt" file to UCSC as a
custom track to view your data.
i.e.
cp PU.1-ChIP-Seq/PU.1-ChIP-Seq.ucsc.bigWig /Web/Server/Root/Path/
** edit PU.1-bigWig.trackInfo.txt to have the right URL **
makeUCSCfile PU.1-ChIP-Seq/ -o PU.1.negativeStrand.bigWig -bigWig chrom.sizes -fsize 1e20 -strand - > PU.1-bigWig.trackInfo.negativeStrand.txt
cp PU.1.positiveStrand.bigWig PU.1.negativeStrand.bigWig /Web/Server/Root/Path/
cat PU.1-bigWig.trackInfo.positiveStrand.txt PU.1-bigWig.trackInfo.negativeStrand.txt > PU.1-bigWig.trackInfo.both.txt
** edit PU.1-bigWig.trackInfo.both.txt to have the right URLs for both the negative and positive strands **
Creating Multi-Experiment Overlay Tracks
To make a "multi-wig hub", as we will refer to them, you need to make sure you have the bedGraphToBigWig program from UCSC, and a working webserver to host your files. If you can handle bigWigs in the section above, you can make multi-wig hubs.
The HOMER program to handle multi-wig hubs is called makeMultiWigHub.pl. It works essentially the same way as the makeBigWig.pl script, however, the syntax is a little different. The basic usage is:
i.e. makeMultiWigHub.pl ES-Factors mm9 -d mES-Oct4/ mES-Sox2/ mES-Nanog/ mES-Klf4/ mES-Esrrb/ mES-cMyc/ mES-Stat3/
NOTE: make sure you use the UCSC genome (e.g. mm9) and not the masked, bastardized HOMER version (mm9r).
The above example will produce a hub called "ES-Factors", composed of configuration files and bigWig files, and place it on your server in the directory specified by "-webDir <directory>". It will also provide you with a URL to the hub (dependent on the value of -url <base url>"). To load the Hub, click on "Track Hubs" on the UCSC browser (next to custom tracks button), and paste the URL in to the dialog box. The example above will look something like this:
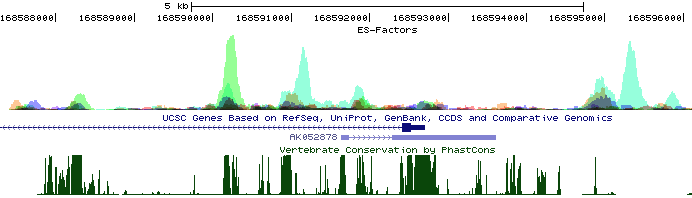
To figure out which factors correspond to which colors, click on the Blue Heading for the Hub in the settings area below the UCSC picture. Something like this should pop up:
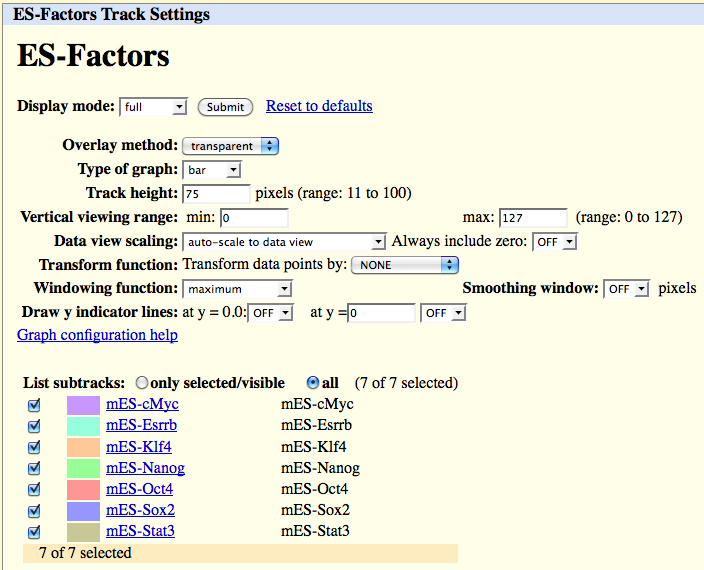
Unfortunately, as of now editing hub information can only be done by directly modifying the hub files on the server. For example, to edit to colors, you must edit the "/webserver/directory/hubName/genome/trackDB.txt" file.
Because Hubs are so cool, HOMER will also do +/- strand RNA data right. Unfortunately, for now you can't mix stranded and non-stranded data in the same hub with the makeMultiWigHub.pl program. To visualize stranded information, add "-strand". Below is an example:
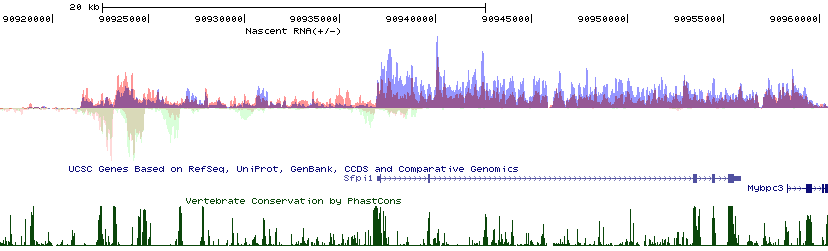
Other makeMultiWigHub.pl options are essentially identical to makeBigWig.pl.
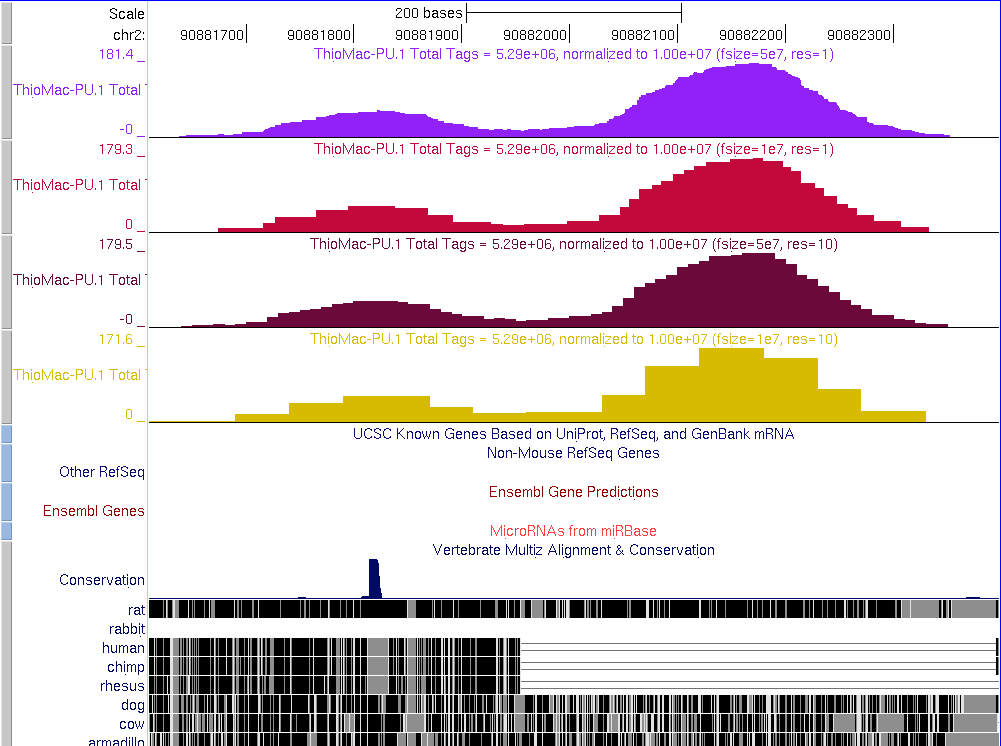
Examples of UCSC bedGraph files
- -fsize 5e7 -res 1
- -fsize 1e7 -res 1
- -fsize 5e7 -res 10
- -fsize 1e7 -res 10
Command line options for makeUCSCfile
Usage: makeUCSCfile <tag directory> [options]Creates a bedgraph file for visualization using the UCSC Genome Browser
General Options:
-fsize <#> (Size of file, when gzipped, default: 1e10, i.e. no reduction)
-strand <both|separate|+|-> (control if reads are separated by strand, default: both)
-fragLength <# | auto | given> (Approximate fragment length, default: auto)
-adjust <#> (Adjust edge of tag 3' by # bp, negative for 5', default: none[good for dnase])
-tbp <#> (Maximum tags per bp to count, default: no limit)
-mintbp <#> (Minimum tags per bp to count, default: no limit)
-res <#> (Resolution, in bp, of file, default: 1)
-avg (report average coverage if resolution is larger than 1bp, default: max is reported)
-lastTag (To keep ucsc happy, last mapped tag is NOT extended by default
Using this option will allow extending of data past the last tag position)
-norm <#> (Total number of tags to normalize experiment to, default: 1e7)
-normLength <#> (Expected length of fragment to normalize to [0=off], default: 100)
-noadj (Do not normalize tag counts)
-neg (plot negative values, i.e. for - strand transcription)
-CpG (Show unmethylated CpG ratios)
-color <(0-255),(0-255),(0-255)> (no spaces, rgb color for UCSC track, default: random)
-i <input tag directory> (normalize bedGraph to input data)
-pseudo <#> (Number of pseudo counts used to smooth out low coverage areas, default: 5)
-log (report log2 ratio instead of linear ratio)
-inputtbp <#>, -inputFragLength <#>, -inputAdjust <#> can also be set
-bigWig <chrom.size file> (creates a full resolution bigWig file and track line file)
This requires bedGraphToBigWig to be available in your executable path
Also, because how how bigWig files work, use "-strand -" and "-strand +"
in separate runs to make strand specific files: "-strand separate" will not work
Consider using makeBigWig.pl and makeMultiWigHub.pl if interested in bigWigs
-o <filename|auto> (send output to this file - will be gzipped, default: prints to stdout)
auto: this will place an appropriately named file in the tag directory
-name <...> (Name of UCSC track, default: auto generated)
-style <option> (See options below:)
chipseq (standard, default)
rnaseq (strand specific, if unstranded add '-strand both' to end of command)
tss (strand specific, single bp fragment length)
dnase (fragments centered on tag position instead of downstream)
methylated (single bp resolution of cytosine methylation)
unmethylated (single bp resolution of unmethylated cytosines)
damid (2kb fragments centered on 5' end of reads)
-circos <chrN:XXX-YYY|genome> (output only a specific region for circos[no header])
Command line options for makeBigWig.pl
Script for automating the process of creating bigWigs
Usage: makeBigWig.pl <tag directory> <genome> [special options] [options]
Special Options for bigWigs [choose one, don't combine]:
-normal (ChIP-Seq style, default)
-strand (Strand specific, for RNA-Seq and GRO-Seq)
-dnase (Special options for Crawford-lab style DNase-Seq)
-cage (Special options for CAGE/TSS-Seq)
-cpg (Special options for mCpG/CpG)
Other options:
Whatever options you want to pass to makeUCSCfile
!!Warning!!: do not try to specify "-strand separate" - use the special option above.
File options:
-fsize <#> (Use to limit the size of the bigwig files)
-url <URL> (URL directory -no filename- to tell UCSC where to look)
-webdir <directory> (name of directory to place resulting bigWig file)
-update (overwrite bigwigs in the webDir directory, otherwise random numbers are
added to make the file unique.
Current url target (-url): http://homer.salk.edu/bigWig/
Current web directory (-webDir): /data/www/bigWig/
You're going to want to modify the $wwwDir and $httpDir variables at the top of
the makeBigWig.pl program file to accomidate your system so you don't have to
specify -url and -webdir all the time.
Command line options for makeMultiWigHub.pl
Script for
automating the process of creating multiWig tracksUsage: makeMultiWigHub.pl <hubname> <genome> [options] -d <tag directory1> [tag directory2]...
Special Options for bigWigs [choose one, don't combine]:
-normal (ChIP-Seq style, default)
-strand (Strand specific, for RNA-Seq and GRO-Seq)
-dnase (Special options for Crawford-lab style DNase-Seq)
-cage (Special options for CAGE/TSS-Seq)
-cpg (Special options for mCpG/CpG)
Other options:
Whatever options you want to pass to makeUCSCfile
!!Warning!!: do not try to specify "-strand separate" - use the special option above.
Also, for the genome, do NOT use repeat version (mm9r) - use mm9 instead
File options:
-force (overwrite existing hub)
-fsize <#> (limit the file size of the bigwig files to this value)
-url <URL> (URL directory -no filename- to tell UCSC where to look)
-webdir <directory> (name of directory to place resulting hub directory)
Current url target (-url): http://biowhat.ucsd.edu/hubs/
Current web directory (-webDir): /data/www/hubs/
You're going to want to modify the $wwwDir and $httpDir variables at the top of
the makeMultiWigHub.pl program file to accomidate your system so you don't have to
specify -url and -webdir all the time.
Next: Finding Peaks (ChIP-enriched
regions) in the genome

Can't figure something out? Questions, comments, concerns, or other feedback:
cbenner@salk.edu