
HOMER
Software for motif discovery and next-sequencing analysis
Selecting Background Sequences (homer2 background)
Traditionally, HOMER has selected background
sequences semi-randomly from the genome, making sure that
the per-region GC% distribution in the background sequences
matched that of the target sequences, explained here.
HOMER2 now offers much greater control of the background
sequence, including routines that help model
position-specific features of background sequence.The new background features are performed using a new subprogram called "homer2 background". This tool allows HOMER to randomly select from the genome or generate synthetic sequences that match positional k-mer content among other things. This means that if your DNA sequences are anchored on a feature of interest, HOMER will attempt to account for lower-order (i.e. dinucleotide) positional sequence bias that might be present in your sequences. For example, if you have sequences centered on transcription start sites (TSS), certain nucleotides are likely to occur at certain positions within your sequences (such as 'CA' near the TSS), which may artificially increase the chance a given DNA motif will be recognized at that position. New background selection methods help address this concern, and provide tools to score the relative enrichment/depletion of motifs as a function of the motif's position in such sequences.
The "homer2 background" program is used by two other utilities, findMotifsGenome.pl and createHomer2EnrichmentTable.pl. However, if you want full control over how HOMER selects background sequences, you can use this tool directly. Furthermore, if you would like to use HOMER to generate background sequences for other applications or for use with other tools and analysis, you may want to use "homer2 background" directly. Ultimately, the output sequences can then be use as controls for DNA motif finding, enrichment calculations, machine learning applications, or other creative uses.
One important note that applies to all of these
approaches - the sequences you analyze must all have the SAME
LENGTH. The assumption for positional
enrichment is that all of your sequences are anchored on
some position or feature of interest such that there is a
relationship between nucleotide 1, 2, 3, etc. across all
of the input sequences. There are no exceptions to
this - if you do have variable length sequences, you [the
user] are in charge of deciding how to trim (or pad them
with N's) to make them the same length.
homer2 background
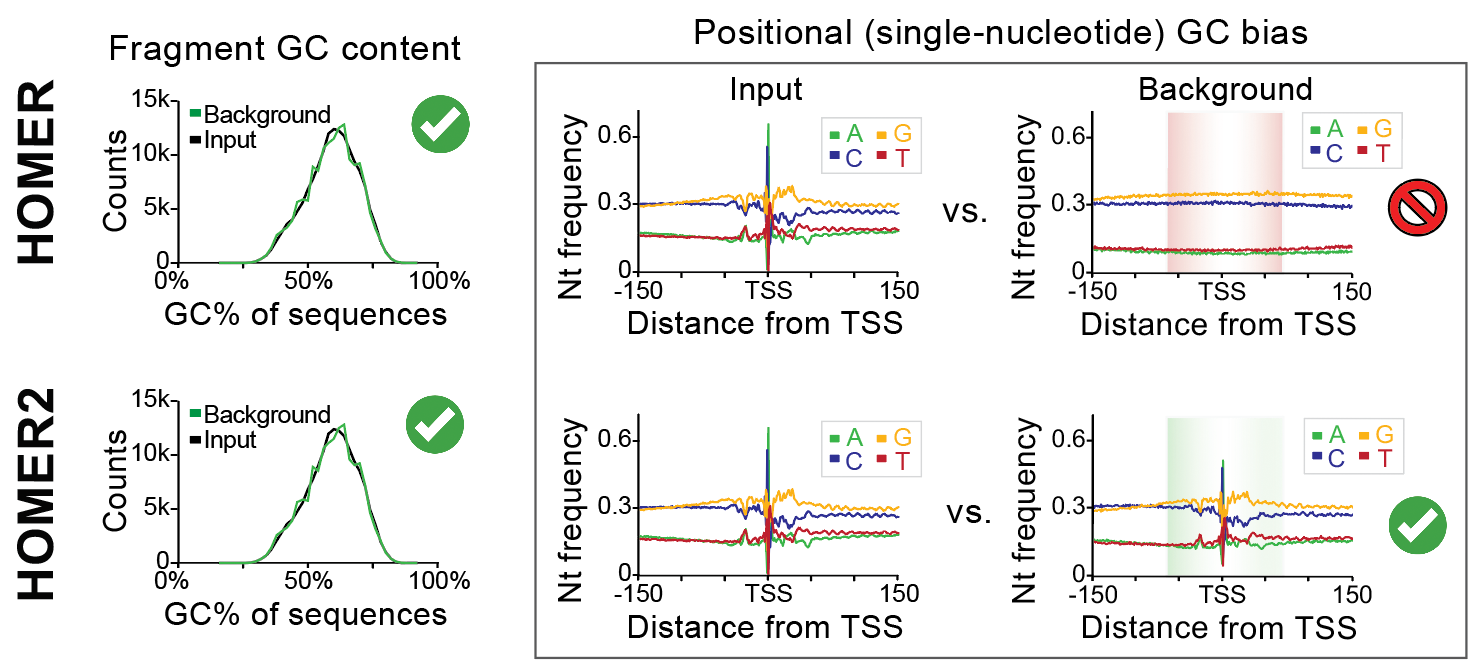
The basic idea is to select background sequences with similar position-specific sequence properties relative to your input/target sequences. Traditionally, HOMER only normalizes for GC content on a per-sequence basis, which helps control for the presence of sequences with extreme levels of GC dinucleotides which are typically found in CpG Islands near vertebrate regulatory regions (e.g. promoters). With HOMER2, not only will HOMER control for the overall per-sequence GC%, it will also attempt to select (or model sequences) that contain similar lower-order nucleotide content (i.e. mononuclotides, dinucleotides, trinucleotides, etc.) at each position.One thing to keep in mind is that this program was designed with regulatory elements in mind. It is meant to work with collections of sequences that are ~50 to 500 bp in length comprised of ~5000 to 500,000 total sequences. Although it can work in other scenarios too, it's behavior and performance may start to falter.
Overview of background selection:
Key inputs and parameters:
- Input sequences (either genomic positions or a FASTA
file, MUST be the same length)
- Genome or other FASTA file to select background sequences from (technically optional as HOMER2 can create synthetic background sequences if desired).
- k = length of k-mer at each position to match sequence properties with (k=1 nucleotides, k=2 dinucleotides, k=3 trinucleotides, etc.)
- Position-dependent or position-independent analysis of k-mer content (i.e. can also normalize k-mer content regardless of position)
- Select actual background sequences from a file or
generate synthetic sequences.
HOMER2 then performs the following steps:
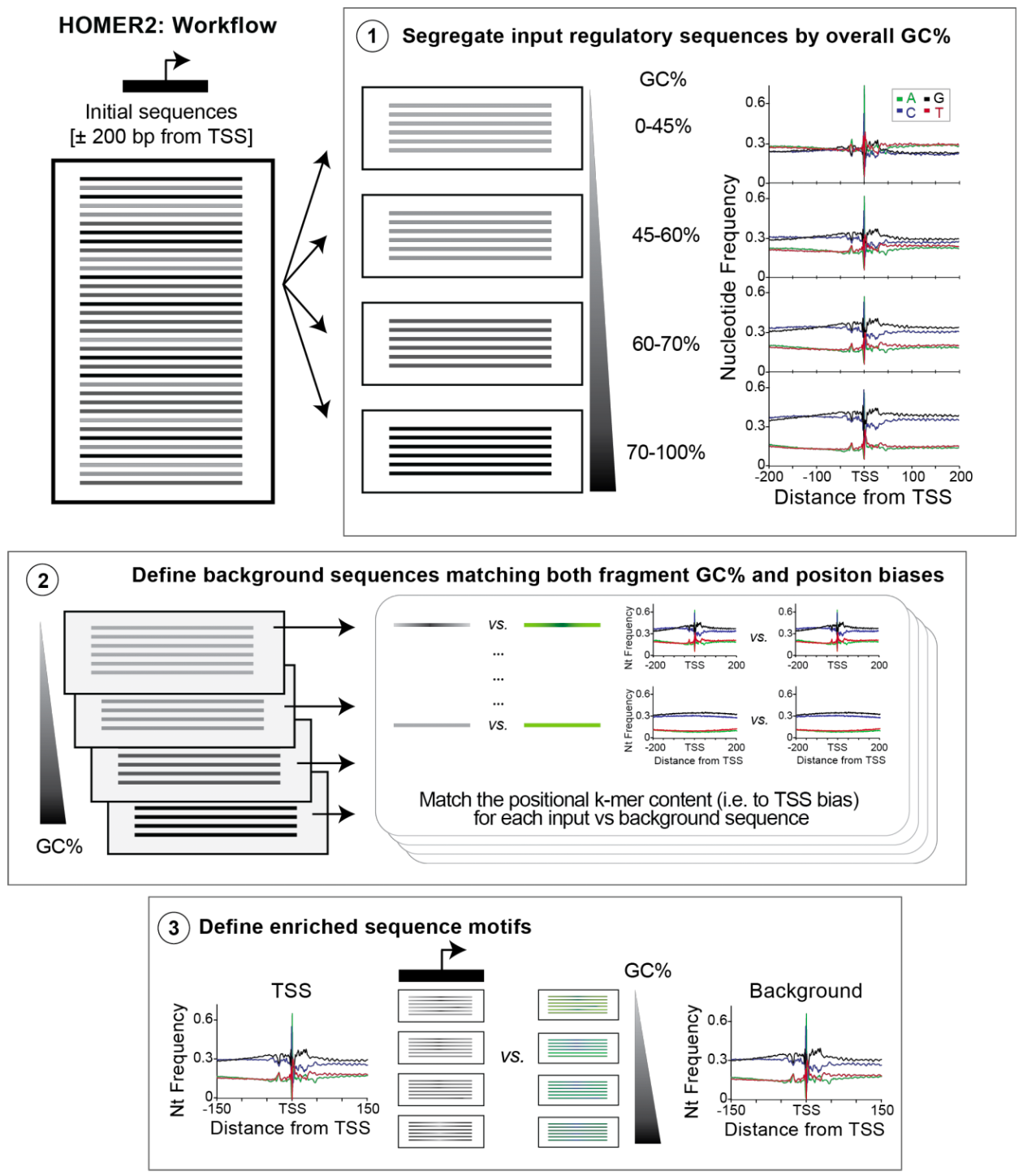
Following the diagram above, (1) HOMER will first segregate your target sequences into 10 equal sized ‘bins’ based on their overall GC content (controlled by the -nbin <#> parameter). This helps yield background sequences with similar overall properties to the target sequences as vertebrate regulatory elements often fall along a spectrum of different overall GC content levels. Otherwise, it is likely the subsequent sequence selection or modeling steps would tend to select background sequences resembling the average GC content of the target set (instead of a mix of extremes), providing a poor set of control sequences. Once it determines the overall GC content of the target sequences, the program will read through the genome (i.e. all possible background sequences) and segregate them into the appropriate GC-bin for background selection at the next step. Since large genomes will cause this assignment to eat up a lot of memory, the relative number of sequences considered at this step can be controlled using the parameter -NN <#> (Default 10 million). The program is ‘smart’ in the sense that when randomly selecting potential background sequences, it will try to select them such that each GC-bin gets a similar number (i.e. if your target sequences are GC rich, it will tend to select GC-rich background sequences at a higher rate relative to AT-rich sequences).
(2) Once candidate background sequences where selected, HOMER will then iteratively select background sequences to use in the final background set that match the positional k-mer content of the targets – doing this separately for each GC-bin. This is done by calculating the average k-mer content at each position for both the target and putative background. The difference between these matrices is calculated and then each target sequences is scored based on summation of the frequency differences at each position based on the k-mer present in the sequence. Sequences are then placed in 10 ‘sub bins’ based on this difference (-nsubBins <#>, instead of overall GC content). The same is done for the putative background sequences, and then a subset of background sequences from each sub bin are then selected to use in the next iteration. This continues until the desired number of background sequences are reached (3).
Sample Data:
- tss.positions.200.txt
- HOMER-style peak file similar to BED that contains
regions centered on 155k TSS locations identified by
csRNA-seq in U2OS cells in human [hg38]). The "200"
refers to the fact that the regions are 200 bp long
(centered on the TSS). Unzip it with gunzip before using
it.
- hg38.fa - you can download this directly from UCSC using "wget https://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/hg38.fa.gz" and then unzipping the file with gunzip
homer2 bg -p tss.positions.200.txt -g hg38.fa -pkmer 2 -N 100000 -NN 100000000 -o output -allowTargetOverlaps -allowBgOverlaps
Output: The program will create several output files starting with <prefix> (set with -o <prefix>, in the case above it is 'output'):
- <prefix>.info.txt - reports the command line options used and some basic information including actual number of sequences in output background set and the cumulative 'difference' or error in the background sequence properties and the target set.
- <prefix>.bg.positions.bed - tab-delimited
BED format file with the genomic positions of all
selected sequences
- <prefix>.bg.sequences.fasta - FASTA
formatted file containing the selected background
sequences
- <prefix>.bg.stats.txt - table containing the name of the background sequence, chromosome, position (min regardless of strand), strand, weight (1.0, not used), Profile Score (internal score, not used), GC%, and the internal bin ID the background was assigned from.
- <prefix>.bg.positions.bed, <prefix>.bg.sequences.fasta,
and <prefix>.target.stats.txt provide the
same information, but for the target sequences.
Key input parameters to consider:
-model
Instead of selecting sequence from the background, the program can also generate synthetic sequences using what is essentially a positional k-mer markov model by adding the parameter "-model" to the command. One advantage is that this is much less resource intensive, and you can essentially generate as many as you want. The downside is that the sequences are synthetic a may not capture some of the higher order sequence features that are nonetheless present in the genome, such as longer simple repeats, etc.k-mer length
By default, homer will attempt to model dinucloetides in a position-dependent manner ("-pkmer 2"). Certain applications might call for longer kmers (i.e. when analyzing coding regions, trinucleotides might be appropriate. However, the higher the k-mer length, the more likely you are to "normalize out" longer signals (such as TF binding sites). Also, the ability of the program to find a background set that matches the desired k-mer positional content exponentially decreases as the length of the k-mer increases. As a useful heuristic, it is best to have at least 100x more target sequences than possible k-mers such that each can be properly quantified. For example, if you want to model trinucleotide content, you probably want 4^3*100=6400 target sequences at minimum.-pkmer <#> vs. -ikmer <#>
To model kmers in a position-dependent manner, use "-pkmer <#>". However, if you don't care about the positional enrichment for each kmer in the dataset, just that overall kmer content of each sequence, you can use "-ikmer <#>" instead. This will generate background sequence sets more akin to traditional motif analysis approaches (including original HOMER).-allowTargetOverlaps and -allowBgOverlaps
When selecting background sequences, the program, by default, will avoid picking sequences that overlap one another. "-allowBgOverlaps" means that the final background sequences that are chosen may overlap some of their bp. By also specifying "-allowTargetOverlaps", it then is possible that some of the background sequences will overlap with the target sequences to some degree (i.e. the sequences should be the same, but some regions may overlap by 10% of their length instead of being completely separate sequences). When performing de novo motif finding in a position independent manner, you probably don't want to allow any overlaps. However, when performing positional enrichment analysis, two overlapping sequences may still contain valuable, independent information even if some of their nucleotides are in common (since their anchor positions are ultimately different), so allowing overlaps could be a good idea. The other practical consideration is that for high GC content regions, there simply aren't very many of them in the genome, and you could simply 'run out' of sequences to chose from if you do not allow some degree of overlap.Practical considerations with respect to performance
-N <#> : Total number of sequences to selectOverall, it might be important to consider that this approach was designed to work with eukaryotic regulatory elements in the size range of 50-500 bp in length. As the k-mer size is increased, or the size of the target regions are increased, the ability of the program to find background sequences that match the properties of the targets will become increasingly difficult and resource intensive, leading to reduced accuracy and long run times.
-NN <#> : Total number of putative background sequences to consider during selection.
Reducing this number can save on memory for big genomes.
-nbins <#> : Number of GC-bins to split the target sequences into (default 10).
-nsubBins <#> : Number of sub bins used when matching positional kmer content (default 10).
Increasing this number can improve accuracy, but again as a useful heuristic, you probably want at least 100-fold more target sequences than the product of these two numbers (i.e. #targets > 10x10x100)
Using the new background selection with other HOMER tools.
DNA motif discovery: findMotifsGenome.pl
Add "-useNewBg" to your command to envoke the new positional dependent background correction. Many of the same parameters above can also be entered on the command line (i.e. -allowBgOverlaps, -pkmer 2)Checking position-dependent motif enrichment for a library of motifs: createHomer2EnrichmentTable.pl
Note: You can always generate background sequences using "homer2 background" and have complete control and then use the resulting FASTA files for motif finding too.
HOMER2 has new routines to help automate the screening of large motif libraries for positional enrichment.
Usage:
homer2 background -i <target sequences.fasta> [options]Generate/Select background sequences that match properties in a set of target sequences.
Inputs:
Target sequences you want to model:
-i <target sequences.fasta> (FASTA file)
-p <target positions.bed> (Alteratively, provide a BED or HOMER peak file with genomic coordinates)
Background sequences to select from:
-model (generate sequences using a model, do not extract real background sequences)
-g <genome.fasta> (genome FASTA file or seqeunce resource to select sequences from)
-b <background sequences.fasta> (explicit set of background sequences to choose from FASTA file)
-bg <background positions.bed> (explicit set of background positions to choose from)
-bgr <background regions positions.bed> (regions of the genome to select bg sequences from)
Key options:
-size <#> (size of regions to consider in background, default: avg of length of target sequences)
-N <#> (number of background sequences to select, default: 100000)
-NN <#> (number of background sequences initial screen from genome, default: 100000000)
-mask (mask lowercase sequence i.e. softmasked sequence, default: use all sequences)
-nbins <#> (number of bins to segregate sequences into for GC selection, def: 10
-nsubBins <#> (number of bins to segregate sequences into for positional frequencies, def: 10
-maxFractionN <#> (Maximum fraction of sequence that can be N and still used, default: 0.5)
-allowTargetOverlaps (allow selected bg sequences from a genome to overlap targets, def: not allowed)
-allowBgOverlaps (allow selected bg sequences from a genome to overlap, def: not allowed)
-strand (allow sequences to overlap if on separate strands)
-pkmer <#> (match positional kmer content)
-ikmer <#> (match overall kmer content [position independent])
-excludeNs/-includeNs (by default, kmers with Ns are excluded when selecting bg sequences,
but included when generating sequences with -model)
-pscore <outputBEDfile> (Report initial pscores)
-maxIterations <#> (maximum iterations, def: 20)
-overlapIteration <#> (iteration to start enforcing no overlaps, def: 5)
-decayRate <#> (selection rate per iteration, def: 0.75)
-seed <#> (seed for random number generator, def: uses time)
Output:
-o <output prefix> (default: out)
-gs (include homer-style group and sequence output files)
Important: You input sequences must be the same length!

Can't figure something out? Questions, comments, concerns, or other feedback:
cbenner@ucsd.edu