|
Practical Tips to Motif Finding with HOMER
Below are some general tips for getting the most out of you
motif
analysis when using HOMER. Be sure to look over this
section about
judging motif quality!
Why is the number of background regions reported by
HOMER different then my input files?
HOMER performs a step to normalize
the GC-content of the background sequences, which
may result in the adjustment of the total apparent
number of background sequences. If you target
sequences are GC-rich and your background sequences are
AT-rich (a common issue with mammalian genomes), many of
the AT-rich sequences may be added fractionally to the
total so that the imbalance is minimized.
Why do motif counts from findMotifsGenome.pl and
annotatePeaks.pl differ?
By default, annotatePeaks.pl uses the given size of the
peaks (default: -size given), while findMotifsGenome.pl
uses a default size of 200 (default: -size 200).
NOTE: New versions require that -size be specified when
using findMotifsGenome.pl...
What to do if motif finding takes too long...
Ctrl+C... If you are using
reasonable parameters (see next section), it shouldn't
take more than
an hour or so, and in most cases much less.
Choosing the length of motifs to find
It's almost always a good
idea to
start with the default parameters. Resist the urge
to find motifs
larger than 12 bp the first time around. Longer
motifs will show
up as different short motifs when finding shorter
motifs. If
there aren't any truly significant motifs when looking at
short motifs,
it is unlikely that you will find good long motifs
either. And it
doesn't take much time to check for short motifs.
i.e. -S 25 -len 8,10,12
Once you do find motifs that look promising, try looking
for longer
motifs.
Finding Long Motifs
The new version of HOMER
(v3.0+)
is better at looking for long motifs. However, it
can be tricky
looking for long motifs because the search space gets very
large.
Also, the running time on longer motifs increases and may
break your
patience.
Since HOMER is an empirical motif finding program, it
starts from
actual oligos present in the sequence and attempts to
figure out if
they are enriched. If you are looking at 20 bp
sequences, there
is a good chance that they are all more-or-less unique in
your data set
with only 1 instance in either the target or background
sequences. HOMER normally allows mismatches in the
original oligo
to see if the oligo together with similar oligos are
collectively
enriched. The problem is that this technique starts
to break down
at long lengths. It takes many mismatches to find
enough related
sequences to assess enrichment, and it is computationally
expensive to
find them.
To maintain sensitivity
for
longer motifs:
Increase the "-mis
<#>"
option to allow more mismatches. In practice, I
would use at
least "-mis 4" or "-mis 5" for sensitive detection of 20
bp
motifs. If the data set is for a strong motif
(i.e. CTCF ChIP-Seq
peaks), then you don't have to worry about this so much
since the motif
signal is very strong.
To find longer version of
a
given motif:
The local optimization
phase
handles long motifs pretty well - long motifs cause more
of a problem
with the global search phase. Usually long motifs
show enrichment
for parts at shorter motif lengths. Another
strategy is to first
find a short version of the motif (i.e. -len 12), and
then rerun HOMER
and tell it to optimize the motif at a longer motif
length with the
"-opt <motif file>". To do this with a motif
named
"motif1.motif":
findMotifsGenome.pl
peaks.txt hg18r OutputDirectory -opt motif1.motif
-len 30
This will enlarge the motif(s) in the motif1.motif to 30
bp and
optimize them.
- try to reduce the number of target sequences to
include
only high quality sequences (such as "focused"
peaks or peak with the highest peak scores).
- try limiting the length of sequences used (i.e. "-size 50" when using
findMotifsGenome.pl)
- try limiting the total number of background sequences
(i.e.
"-N 20000" when
using findMotifsGenome.pl)
In a practical sense, you
should
be able to search for motifs of length
20 or 30 when analyzing ~10k peaks with parameters "-len
20,30 -size 50
-N 25000 -mis 5". HOMER wasn't really designed to
find really
long
motifs; since it is an empirical motif finder, the
sequence "space"
gets a bit sparse at lengths >16, but in practice it
still works.
How many sequences can HOMER handle?
In theory, a lot (i.e.
millions). It has been designed to work well with
~10k target
sequences and 50k background sequences. If you are
using a large
number of sequences with findMotifs.pl,
you
many
want
to
use
the
" -b"
option, which switches to the cumulative
binomial
distribution for motif scoring, which is faster to
calculate and gives essentially the same results when
using large
numbers of sequences. The binomial is used by
default in findMotifsGenome.pl.
(I guess it
should be called BOMER !?).
Choosing background sequences
Most of the methods in HOMER
attempt to select the proper background for you, but in
some cases this
doesn't work. Normally, HOMER attempts to normalize
the GC
content in target and background sequences. If you
believe
normalizing the CpG content is better, use the option " -cpg" when performing
motif finding
with either findMotifs.pl
or findMotifsGenome.pl.
In some cases the user may have a better idea of what the
background
should be, so HOMER offers the following options:
Promoters:
When using
analyzing promoters with
findMotifs.pl,
if
you
wish
to
use
a
specific set of promoters as background, place
them in a text file (1st column is the ID) and use the " -bg <background IDs
file>"
option. Genes found in the target and background
will be removed
from the background set so that they don't cancel out
each other.
Examples:
- Use expressed genes from a microarray as
background
- Use only genes represented on the microarray as
background
Genomic
Regions:
When analyzing peaks/regions with findMotifsGenome.pl,
you
can
specify
the
genomic
regions
of appropriate background regions
by placing them in their own peak file and using the " -bg <background peak
file>".
Examples:
- Specify peaks common to two cell types as
background when
trying to find motifs specific to a set of cell-type
specific peaks -
this will help cancel out the primary motif and
reveal the co-enriched
motifs
- If peaks are near Exons, specify regions on Exons
as
background to remove triplet bias.
FASTA Files:
Here you have (the necessary) freedom to specify
whatever you want!
Please note, that if the
number
of background sequences is small, or similar in number to
the number of
target sequences, you should consider switching to the
hypergeometric
distribution to improve accuracy when using findMotifsGenome.pl ("-h").
You man also want to disable
CpG/GC normalization depending on how you selected your
background,
which can be done with "-noweight".
Sequence Bias, GC/CpG normalization, and
Autonormalization
Be default, homer performs
several normalization steps to make sure the sequences
that are being
analyzed look reasonable (details here).
Since
GC% differences are the largest source of bias, these are
dealt
with during the background selection stage to minimize any
issues.
Other types of sequence bias may be present in your
data. The
purpose of the autonormalization routines (" -nlen <#>" and " -olen <#>") are
there to help
deal with this type of bias. If your results have
strong
enrichment for simple nucleotide repeats, you may want to
try "- olen <#>"
which will more
aggressively normalize the data.
How
to
Judge the Quality of the Motifs Found
WARNING: Because this is the hardest
thing for people to understand, I'll say it again
here. HOMER
will print the best guess for the motif next to the
motif results, but
before you tell your adviser that your factor is
enriched for that
motif, it is highly recommended that you look at the
alignment!!!
Here
is
an
example
of
what might be going on:

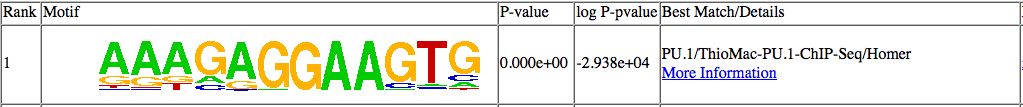
In this case, HOMER has identified YY1 as the "best
guess" match for
this de novo
motif.
Well, lets click on "More Information" and see what's
up:
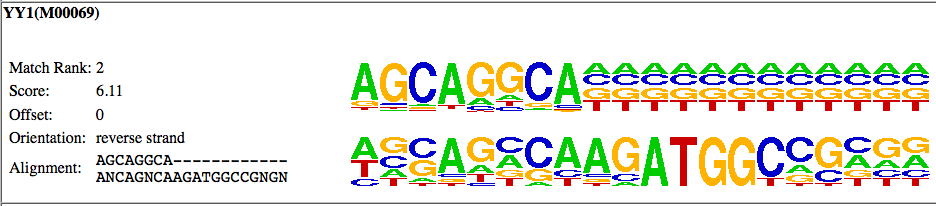
As you can see in this case, the motif aligns to the
edge of the known
YY1 motif, and not to the core of the YY1 motif
(CAAGATGGC). This
doesn't mean that the YY1 motif is not enriched in your
data, but
unless there are other motif results that show
enrichment of the other
parts of the YY1 motif, it is not likely that the YY1
motif is enriched
in your data set.
And as always,
remember that
HOMER is a de novo motif tool!!!
Even though HOMER
will guess the best match, if it is a novel motif,
your don't want to
trust that match anyway. Hence, the you can see
the importance of
viewing the alignment and getting a feel for what
evidence exists
either for or against this assignment.
There are many cases where HOMER will find motifs with
very low
p-values, but the motifs might look "suspicious".
Poor quality
motifs can be loosely classified into the following
groups:
Low Complexity
Motifs:
(less of a problem with
the
v3.0+) These types of motifs tend to
show preference for same collection of 1, 2, 3, or 4
nucleotides in
each position and are typically very degenerate.
For example:
These motifs typically arise when a systematic bias
exists between
target and background sequence sets. Commonly
they will be very
high in GC-content, in which case you may want to try
adding " -gc"
to your motif finding command
to normalize by total GC-content instead of
CpG-content.
Other times this will come up when analyzing sequences
for various
genomic features that have not been controlled for in
the background -
for example, comparing sequences from promoters to
random genomic
background sequences in some organisms will show
preferences for
purines or pyrimidines. HOMER is very sensitive,
so if there is a
bias in the composition of the sequences, HOMER will
likely pick it
up. Autonormalization in the new version
minimizes this problem.
Simple Repeat
Motifs:
(less of a problem with
the
v3.0+) Some times motifs will show
repeats of certain patterns:
Usually motifs like this will be accompanied by several
other motifs
looking highly similar. Unless there is a good
reason to believe
these may be real, it's best to assume there is likely a
problem with
the background. These can arise if your target
sequences are
highly enriched on exons (think triplets) and other
types of sequences,
and if " -gc"
doesn't help, you
may have to think hard about the types of sequences that
you are trying
to analyze and try to match them. (i.e. Promoters
vs. Promoters,
Exons vs. Exons etc.) You can also try upping the
ante by using " -olen
<#>" to autonormalize
sequence bias at the oligo level.
Small Quantity
Motifs / Repeats:
These are a little
harder to
explain. These look like real motifs but are
found in an
incredibly low percentage of targets - i.e. like an
oligo or part of a
repeat that is in a couple of the target sequences
that appears as a
significant motif. Statistically speaking they
are enriched, but
likely not real. These are the biggest problem
when looking for
motifs in promoters from a small list of regulated
genes. In
principle, in a motif is present in less
than
5%
of
the
targets
sequences, there may be a problem.
Leftover Junk:
These are motifs that
appear in
your lower in your results list after you've
discovered high quality
motifs. If an element is highly enriched in your
sequences, HOMER
will (hopefully) find it, mask it, and then continue
to look for
motifs. In this case, many of the other motifs
that HOMER finds
will be offsets or degenerate versions of highly
enriched motif(s)
found
at the beginning. For example (another PU.1
example):
This are not necessarily negative results, but they
should be place in
context. This commonly happens in ChIP-Seq data
sets where the
immunoprecipitated protein is highly expressed and
binds strongly a ton
of binding sites. These "other" motifs are
likely also capable of
binding PU.1 and probably represent low affinity
binding sites, but
giving them too much individual attention is not
recommended in this
context given they are motifs that have been
constructed using leftover
oligos in the motif finding process that didn't make
it into the most
highly enrichment motifs. A safer way to
approach these elements
is to repeat the motif finding procedure with regions
lacking the top
motif, or by adding " -mask
<motif
file>" to the motif finding command to
cleanly mask the top
motif from the motif finding procedure.
|