
HOMER
Software for motif discovery and next-gen sequencing analysis
Initial Hi-C data processing with HOMER
This page describes the general workflow for processing
FASTQ files from Hi-C to use with HOMER. The general
idea is to trim the FASTQ files, align them to the reference
genome, and then assembly them into a HOMER-style tag
directory. Below is a quick reference that describes
that commands that most researchers will use to process a
Hi-C experiment, followed by a more detailed description of
the options available with HOMER and a discussion of
potential parameters that one might want to change.Quick Reference:
#FASTQ trimming and read alignment:
#reads should be trimmed and aligned separately (do not perform paired-end alignment with HOMER). Assumes MboI/DpnII (GATC) is the restriction enzyme used in the Hi-C assay:
homerTools trim -3 GATC -mis 0 -matchStart 20 -min 20 hicExp1_R1_fastq
homerTools trim -3 GATC -mis 0 -matchStart 20 -min 20 hicExp1_R2_fastq
bowtie2 -p 20 -x hg38index -U hicExp1_R1_fastq.trimmed > hicExp1_R1.hg38.sam
bowtie2 -p 20 -x hg38index -U hicExp1_R2_fastq.trimmed > hicExp1_R2.hg38.sam
#Create Hi-C Tag Directory with HOMER:
#Paired alignment files should be provided with a comma (NO spaces around the comma). The "-tbp 1" removes PCR duplicates and is highly recommended:
makeTagDirectory HicExp1TagDir/ hicExp1_R1.hg38.sam,hicExp1_R2.hg38.sam -tbp 1
#optional - for more thorough QC read-outs (takes longer):
makeTagDirectory HicExp1TagDir/ hicExp1_R1.hg38.sam,hicExp1_R2.hg38.sam -tbp 1 -genome hg38 -checkGC -restrictionSite GATC
#optional - create a *.hic file if you have juicer_tools installed to visualize with Juicebox (output file will be placed inside the tag directory):
tagDir2hicFile.pl HicExp1TagDir/ -juicer auto -genome hg38 -p 10
Full Reference:
Trimming FASTQ files from Hi-C experiments
Hi-C and variant technologies use paired-end sequencing to assess regions of the genome that are cross-linked in the same spatial location. After sequencing, you should have two files (or many sets of paired files) for the first and second reads from each read pair. Unlike paired end sequencing for other techniques like genomic resequencing or RNA-Seq, where you might expect the 2nd read to be located within the general vicinity of the first read, read-pairs from Hi-C experiments should be processed independently.
First step for processing Hi-C FASTQ files is to remove sequences that might come from the proximity ligation of other regions of the genome to the DNA fragment. These can complicate alignment since [together with the beginning of the read] they do not represent a continuous genomic sequence. To improve mapping rates, it's best to trim the sequences at the restriction site. You can use whatever tool that you want to use for this, here we'll use homerTools (more info on homerTools):
If using a standard 4-cutter like MboI or DpnII (GATC):
homerTools trim -3 GATC -mis 0 -matchStart 20 -min 20 hicExp1_R1_fastqIf using a restriction enzyme that produces different types of overhangs, like the 6-cutter HindIII, the fill in step can create sequences that differ from the actual restriction site (AAGCTT becomes AAGCTAGCTT):
homerTools trim -3 AAGCTAGCTT -mis 0 -matchStart 20 -min 20 hicExp1_R1_fastqBoth of these commands will produce an output file with "*.trimmed" at the end (e.g. "hicExp1_R1_fastq.trimmed"). These commands also work on gzipped FASTQ files. Be sure to trim both the 1st and 2nd reads.
Aligning Hi-C Experiments to the Reference Genome
After trimming the FASTQ files, the next step is to align them to the reference genome. It is highly recommended to align to a reference genome composed of only canonical chromosomes - do not include *_random.fa scaffolds or other chromosomal fragments since they have not been properly assembled and will only introduce noise into the Hi-C analysis. We recommend aligning the trimmed FASTQ reads with either bwa or bowtie2, although any modern short read aligner should work fine. Be sure to align each read separately - do not align the reads in paired-end mode!
bowtie2 -p 20 -x hg38index -U hicExp1_R1_fastq.trimmed > hicExp1_R1.hg38.samHi-C experiments should produce alignment rates that are comparable to other genomic DNA assays like ChIP-seq, ATAC-seq, or genomic sequencing in general (i.e. >90%). Depending on the organism and read length you may have a different percentage of multi-mappers, but again, these numbers should be in the range of normal DNA sequencing.
bowtie2 -p 20 -x hg38index -U hicExp1_R2_fastq.trimmed > hicExp1_R2.hg38.sam
NOTE: HOMER cannot be used for Hi-C analysis with 1000+ chromosomes per genome (Hi-C analysis is not compatible with the '-single' flag).
Creating a Paired-End Tag Directory with HOMER
Once Hi-C reads have been
mapped, the makeTagDirectory
program is used to process the alignment file into a
HOMER-style tag directory which is used for subsequent
analysis steps. This step is important and is used to
setup some important parameters for analysis, including
the filtering of read-pairs that contribute to noise in
the Hi-C analysis. The basic approach described below
works well for most modern in situ Hi-C experiments (using
4-cutters), but a more detailed description of options is
available further down the page.
To create a paired-end tag directory, you must run makeTagDirectory by entering the two halves of the mapped reads separated by a comma, without spaces. For example:
HOMER matches reads between alignment files by matching the read names in the first column of the SAM file. One potential problem is that the alignment program may encode the read names such that they cannot be easily matched by HOMER. For example, the original bowtie alignment files would leave a 0 or 1 at the end of the read names originating from either the first or second read in the read pair. To work with these files, the program has a special option ("-bowtiePE") which instructs the program to drop the last character from the read names when trying to match them.
The one option that I would highly recommend always adding to the makeTagDirectory command for Hi-C data is "-tbp 1", which will only consider read pairs with the exact same ends once (i.e. remove all PCR duplicates). Given the Hi-C protocol, you would rarely expect to see reads-pairs start from the exact same places twice in the data (at least in the case of libraries generated with a sonication step). If you do, they are likely clonal and a result of over-sequencing your library or some other amplification artifact. This may be different for libraries sequenced to extreme depths.
On a side note, the HOMER tag directory created from Hi-C reads is very similar to the directories created when running makeTagDirectory for other experiments. The paired end reads are stored in *.tags.tsv files (extended format to include the pair), only each read pair will be recorded twice in the appropriate chromosome *.tags.tsv files so that the reads can be indexed and referenced regardless of which chromosome is being analyzed. A flag is also set in the tagInfo.txt file to inform HOMER to treat the experiment as a paired-end Hi-C file. Many of the programs in HOMER, such as makeUCSCfile, will treat paired-end tag directories as "single-end" directories to create output. This can be useful if you want to visualize read coverage in the UCSC Genome Browser or want to find peaks in the Hi-C read coverage with findPeaks, even though these operations are not necessarily well defined scientifically.
One important note: the tagInfo.txt file will report the "Total Tags" value as 2x the total number interactions. This is because HOMER is storing each PE tag twice - the software later adjusts this number to reflect this redundancy, but keep this in mind when interpreting this number.
To create a paired-end tag directory, you must run makeTagDirectory by entering the two halves of the mapped reads separated by a comma, without spaces. For example:
makeTagDirectory <OutputTagDirectory>
<alignment file-read1>,<alignment
file-read2> ... [-tbp 1]
i.e. makeTagDirectory ES-HiC lane1-read1.sam,lane1-read2.sam lane2-read1.sam,lane2-read2.sam -tbp 1
i.e. makeTagDirectory ES-HiC lane1-read1.sam,lane1-read2.sam lane2-read1.sam,lane2-read2.sam -tbp 1
HOMER matches reads between alignment files by matching the read names in the first column of the SAM file. One potential problem is that the alignment program may encode the read names such that they cannot be easily matched by HOMER. For example, the original bowtie alignment files would leave a 0 or 1 at the end of the read names originating from either the first or second read in the read pair. To work with these files, the program has a special option ("-bowtiePE") which instructs the program to drop the last character from the read names when trying to match them.
The one option that I would highly recommend always adding to the makeTagDirectory command for Hi-C data is "-tbp 1", which will only consider read pairs with the exact same ends once (i.e. remove all PCR duplicates). Given the Hi-C protocol, you would rarely expect to see reads-pairs start from the exact same places twice in the data (at least in the case of libraries generated with a sonication step). If you do, they are likely clonal and a result of over-sequencing your library or some other amplification artifact. This may be different for libraries sequenced to extreme depths.
On a side note, the HOMER tag directory created from Hi-C reads is very similar to the directories created when running makeTagDirectory for other experiments. The paired end reads are stored in *.tags.tsv files (extended format to include the pair), only each read pair will be recorded twice in the appropriate chromosome *.tags.tsv files so that the reads can be indexed and referenced regardless of which chromosome is being analyzed. A flag is also set in the tagInfo.txt file to inform HOMER to treat the experiment as a paired-end Hi-C file. Many of the programs in HOMER, such as makeUCSCfile, will treat paired-end tag directories as "single-end" directories to create output. This can be useful if you want to visualize read coverage in the UCSC Genome Browser or want to find peaks in the Hi-C read coverage with findPeaks, even though these operations are not necessarily well defined scientifically.
One important note: the tagInfo.txt file will report the "Total Tags" value as 2x the total number interactions. This is because HOMER is storing each PE tag twice - the software later adjusts this number to reflect this redundancy, but keep this in mind when interpreting this number.
Alternative Input Files
HOMER will also accept "Hi-C Summary" files (use "-format HiCsummary"), which is a simple text file format that contains the mapping positions for a read-pair on each line. These files are tab-delimited text files with the following column assignments:
Hi-C Summary Format (columns):An example of using a Hi-C summary file:
1. Read Name (can be blank)
2. chromosome for read 1
3. positions for read 1 (5' end of read, one-indexed)
4. strand of read 1 (+ or -)
5. chromosome for read 2
6. positions for read 2 (5' end of read, one-indexed)
7. strand of read 2 (+ or -)
makeTagDirectory proB-HiC -format HiCsummary proB.HiC.FA.summary.txt -tbp 1You can also combine Hi-C tag directories or add *tags.tsv files, just like with the regular makeTagDirectory command. For example, to combine directories:
makeTagDirectory ES-HiC-All -d ES-HiC-rep1/ ES-HiC-rep2/ ES-HiC-rep3/ -tbp 1
Hi-C Quality Control
When running the makeTagDirectory
program, several standard quality control analysis files
will be created that resemble the analysis carried out for
single-end reads. The program will also perform some Hi-C
specific quality control checks, including calculating the
percentage of interchromosomal reads or "local
interactions" (i.e. interactions representing religation
to the same site or DNA fragment noise in the sample). You
can also check for GC bias by specifying the genome (i.e.
"-genome hg19")
and "-checkGC" at
the command line (analysis will be conducted by treating
the samples as "single" reads). You can also map the
distribution of reads relative to restriction sites by
using the "-restrictionSite
<seq>" option described below.
The following two Hi-C/paired-end read specific QC files will also be produced by default:
petagLocalDistribution.txt
The following two Hi-C/paired-end read specific QC files will also be produced by default:
petagLocalDistribution.txt
This file is similar to
the autocorrelation file produced with ChIP-seq
experiments, except that it shows the relationship
between the 5' ends of the paired reads (instead of the
general relationship between all tags). This data
is used to determine the fragment size of the Hi-C
fragments used for sequencing, assuming that we should
see a natural peak from "re-ligation events" or random
genomic fragments that got into the sequencing
library. Below is an example (Here we'd
estimate the fragment length to be about 350 bp):
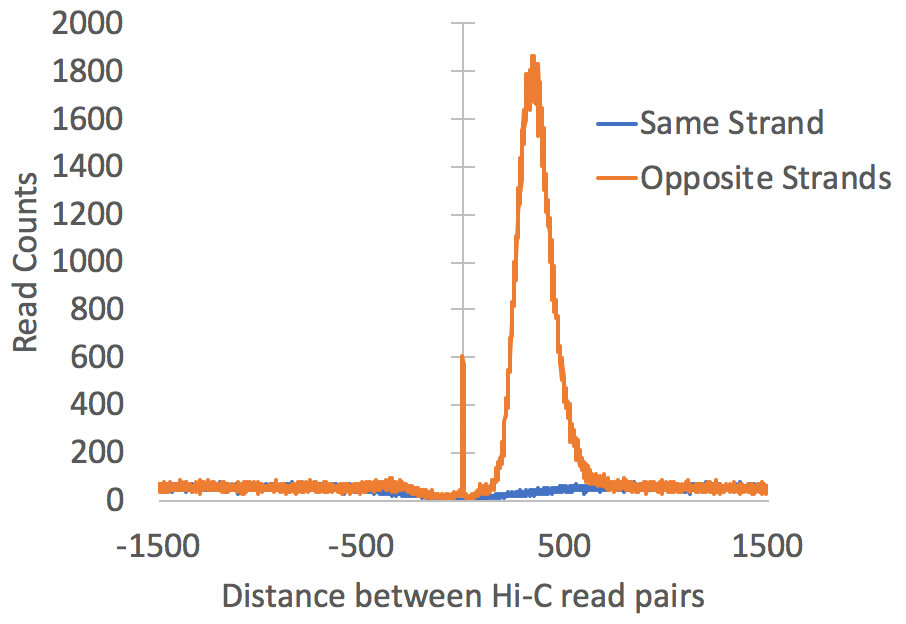
petagDistDistribution.txt
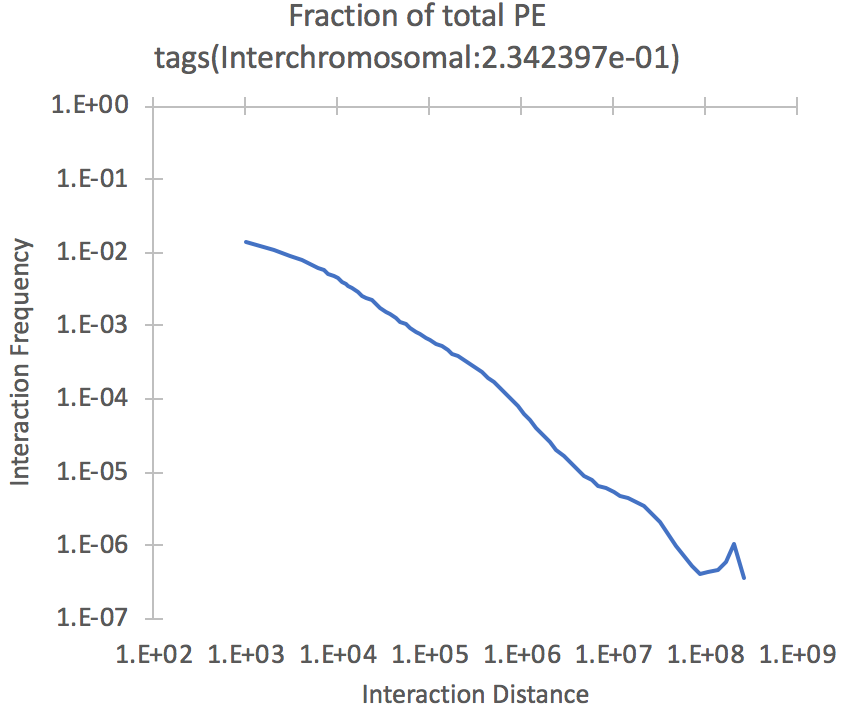
This file contains a
histogram describing the fraction of paired-end reads
that are found at different distances from one
another. The file stops at 300 million bp, and is
divided into 1kb bins. The top of the file also
contains the fraction of interchromosomal
interactions. Below is an example (plotted as a
log vs. log plot). The fraction of PE reads with
interactions greater than 300 Mb in distance are shown
in the final point.
Restriction Sites
One major problem that can arise during Hi-C is that "background" ligation events may occur. These are ligation events that occur after sonication [I think] as they seem to occur between random fragments of DNA with no regard to restriction site used for the Hi-C assay (problems with this may differ between protocols). In theory, all reads should be in the vicinity of the restriction site used for the assay, and if they aren't, there is a good chance they are noise (more later on this). To see the distribution of reads around your restriction site, use the option "-restrictionSite <seq>" (i.e. "-restrictionSite AAGCTT"). You MUST specify the genome so that HOMER can figure out where the restriction sites are located (i.e. "-genome mm9"). If the restriction site has a lot of star activity, you can also add "-rsmis <#>" (i.e. "-rsmis 1") to indicate how many mismatches are tolerated in the sites. By default it only looks for perfect restriction sites. This is much less of a problem (and harder to address) with 4-cutter enzyme restriction sites as are used in most modern protocols. Also note that for 4-cutters, Hi-C fragments often start at the restriction sites (strong spikes in signal near 0).
Adding these options will produce a file named "petagRestrictionDistribution.<seq>.mis<#>.txt" (such as petagRestrictionDistribution.AAGCTT.mis0.txt). This file will display the distribution of reads relative to the restriction site. Graphing it with Excel will look something like this:
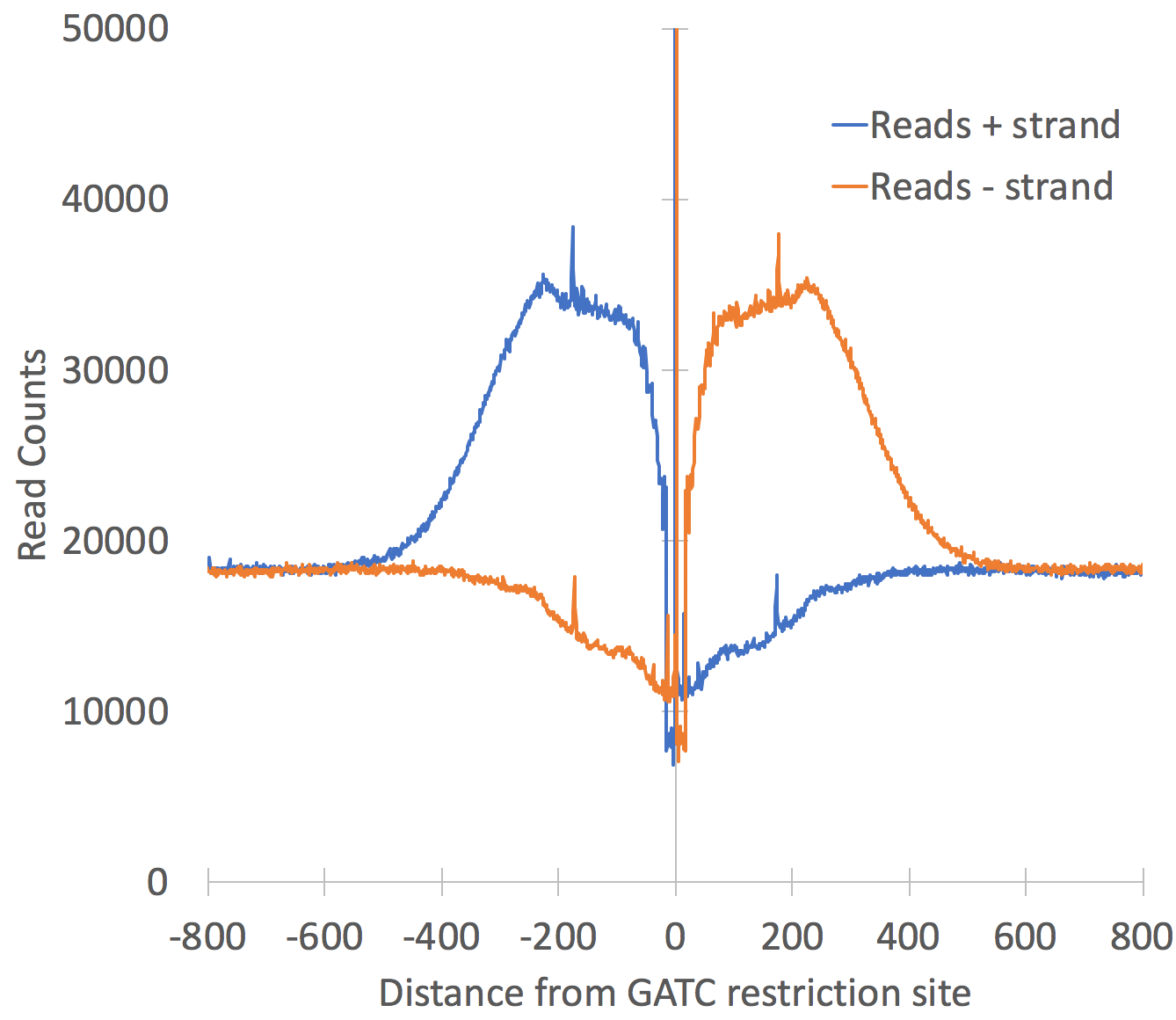
Evaluating Hi-C Quality Controls:
When evaluating Hi-C data quality we would recommend paying attention to the following:
- Genomic Alignment Rate - The number of reads that align to the genome should be similar to that of a ChIP-seq or DNA sequencing experiment for your given organism. In addition, the percentage of multimappers should be similar to those experiments as well. (Mouse/Human experiments should generally have a >90% mapping rate and no more than 20-30% multimappers). If your numbers are close to this you shouldn't have a problem, but if the multimappers represent >50% of the alignments it's likely there is a problem.
- Clonality/PCR duplicates - If the median number of read-pairs per positions is greater than 1, it is likely that the complexity of the library is low. These experiments should not be sequenced more, and in some cases may need to be repeated.
- Percentage of local interactions - Local interactions (<1kb) should compose a relatively small percentage of the total reads. A high percentage of local interactions likely arises from random DNA fragments in the library that do not represent true Hi-C interactions. (If more than 10-20% of reads should be under 1kb apart, and in ideal cases this number should be even lower). If this number changes a lot between experiments, consider removing local interactions using "-removePEbg" explained below.
- Percentage of interchromosomal interactions - A high percentage of interchromosomal interactions may indicate that the Hi-C library is picking up non-specific interactions between regions. Most in situ Hi-C experiments yield interchromosomal interactions rates in the 10-30% range for mammalian samples.
- Contact frequency vs. distance - The relationship between distance and interaction frequency should look roughly linear in a log-log plot as shown above for the petagDistDistribution.txt plot. If it doesn't, there may be a problem with the Hi-C library.
Filtering Uninformative Reads
The Hi-C protocol can
produce sequencing libraries that contain many read pairs
that are not representative of true interactions.
This includes continuous genomic fragments, self-ligation
or re-ligation products, and background non-specific
ligation that occurs independent of a restriction site.
** Modern Hi-C protocols like in situ Hi-C do not suffer from as many of these problems as earlier data, and in many cases you can skip this section if your experiment passes the basic quality controls specified above.
-tbp 1 : (Removes Clonal/PCR duplicates). This is the one filtering step recommended for ALL Hi-C experiments. Please not that if the experiment has high clonality rates (medianReadsPerPosition > 1 in the tagInfo.txt file), then it is not recommended to sequence the library any more and extra scrutiny should be applied when considering the overall experiment quality.
-removePEbg : (Removes local interactions) This will remove paired-end reads that are likely continuous genomic fragments or re-ligation events. If this option is specified, read pairs are removed if they are separated less than 1.5x the sequencing insert fragment length. This distance is estimated from the distribution found in the file "petagLocalDistribution.txt" described above. If you wish to specify your own size, use "-fragLength <#>" to set this length manually.
-removeSpikes <#size> <# fold> : (Removes regions with abnormally high concentrations in reads) This option will remove PE reads that originate from regions of unusually high tag density. These regions arise for various reasons, for example, a region was duplicated in a given cell line. These types of anomalies tend to create artifacts in the data. Removing these from the analysis can help clean things up. The size parameter is the window of tag density that will be examined to find anomalies, and #-fold is how many tags over the genomic average is needed to determine if a locus should be removed from the dataset. I'd recommend something like "-removeSpikes 10000 5" to get rid of the regions that are really bad. For example, this will scan all 10kb regions in the genome, calculate the average read density, and remove reads from regions that contain more than 5x the average number of reads.
** Modern Hi-C protocols like in situ Hi-C do not suffer from as many of these problems as earlier data, and in many cases you can skip this section if your experiment passes the basic quality controls specified above.
-tbp 1 : (Removes Clonal/PCR duplicates). This is the one filtering step recommended for ALL Hi-C experiments. Please not that if the experiment has high clonality rates (medianReadsPerPosition > 1 in the tagInfo.txt file), then it is not recommended to sequence the library any more and extra scrutiny should be applied when considering the overall experiment quality.
-removePEbg : (Removes local interactions) This will remove paired-end reads that are likely continuous genomic fragments or re-ligation events. If this option is specified, read pairs are removed if they are separated less than 1.5x the sequencing insert fragment length. This distance is estimated from the distribution found in the file "petagLocalDistribution.txt" described above. If you wish to specify your own size, use "-fragLength <#>" to set this length manually.
-removeSpikes <#size> <# fold> : (Removes regions with abnormally high concentrations in reads) This option will remove PE reads that originate from regions of unusually high tag density. These regions arise for various reasons, for example, a region was duplicated in a given cell line. These types of anomalies tend to create artifacts in the data. Removing these from the analysis can help clean things up. The size parameter is the window of tag density that will be examined to find anomalies, and #-fold is how many tags over the genomic average is needed to determine if a locus should be removed from the dataset. I'd recommend something like "-removeSpikes 10000 5" to get rid of the regions that are really bad. For example, this will scan all 10kb regions in the genome, calculate the average read density, and remove reads from regions that contain more than 5x the average number of reads.
Filtering Reads based on Restriction Sites
(Only really useful for
experiments using 6-cutter enzymes like HindIII - it is
much more difficult to distinguish these noise patterns
in 4-cutter data since the density of restriction sites
is so high)
All the options in this section depend on restriction sites and require the following to be part of the command:
-restrictionSite <seq> -genome <genome>
Restriction Site Proximity: The following options can be used to specify which reads should be kept with respect to the location of nearby restriction sites. The distance used to assign reads to restriction site is 1.5x the fragment length by default (set it with -restrictionSiteLength <#>):
-removeSelfLigation : Remove reads if their ends form a self ligation with adjacent restriction sites.
-removeRestrictionEnds : Removes reads if one of their ends starts on a restriction site.
-restrictionSiteLength <#> : Maximum distance to a restriction site for assignment.
All the options in this section depend on restriction sites and require the following to be part of the command:
-restrictionSite <seq> -genome <genome>
Restriction Site Proximity: The following options can be used to specify which reads should be kept with respect to the location of nearby restriction sites. The distance used to assign reads to restriction site is 1.5x the fragment length by default (set it with -restrictionSiteLength <#>):
-both : Only keep reads if both ends of the paired-end read have a restriction site within the fragment length estimate 3' to the read. This is the most conservative, and probably recommended for most types of analysis.
-one : Only keep reads if one or both ends of the paired-end read are near restriction sites.
-onlyOne : Only keep reads if one (and only one) of the reads in a paired-end read are near a restriction sites (mainly used for troubleshooting).
-none : Only keep read if neither of the ends are near a restriction site (mainly used for troubleshooting)
-removeSelfLigation : Remove reads if their ends form a self ligation with adjacent restriction sites.
-removeRestrictionEnds : Removes reads if one of their ends starts on a restriction site.
-restrictionSiteLength <#> : Maximum distance to a restriction site for assignment.
Command Line Options for makeTagDirectory:
Usage: makeTagDirectory <directory> <alignment file 1> [file 2] ... [options]
Creates a platform-independent 'tag directory' for later analysis.
Currently BED, eland, bowtie, and sam files are accepted. The program will try to
automatically detect the alignment format if not specified. Program will also
unzip *.gz, *.bz2, and *.zip files and convert *.bam to sam files on the fly
Existing tag directories can be added or combined to make a new one using -d/-t
If more than one format is needed and the program cannot auto-detect it properly,
make separate tag directories by running the program separately, then combine them.
To perform QC/manipulations on an existing tag directory, add "-update"
Options:
-fragLength <# | given | pe> (Set estimated fragment length or use PE length - given: use read lengths)
By default treats the sample as a single read ChIP-Seq experiment
-format <X> where X can be: (with column specifications underneath)
bed - BED format files:
(1:chr,2:start,3:end,4:+/- or read name,5:# tags,6:+/-)
-force5th (5th column of BED file contains # of reads mapping to position)
sam - SAM formatted files (use samTools to covert BAMs into SAM if you have BAM)
-unique (keep if there is a single best alignment based on mapq)
-mapq <#> (Minimum mapq for -unique, default: 10, set negative to use AS:i:/XS:i:)
-keepOne (keep one of the best alignments even if others exist)
-keepAll (include all alignments in SAM file)
-mis (Maximum allowed mismatches, default: no limit, uses MD:Z: tag)
-sspe (strand specific, paired-end reads[flips strand of 2nd read to match])
-read1/-read2 (only analyze 1st or 2nd read for PE sequencing)
bowtie - output from bowtie (run with --best -k 2 options)
(1:read name,2:+/-,3:chr,4:position,5:seq,6:quality,7:NA,8:misInfo)
eland_result - output from basic eland
(1:read name,2:seq,3:code,4:#zeroMM,5:#oneMM,6:#twoMM,7:chr,
8:position,9:F/R,10-:mismatches
eland_export - output from illumina pipeline (22 columns total)
(1-5:read name info,9:sequence,10:quality,11:chr,13:position,14:strand)
eland_extended - output from illumina pipeline (4 columns total)
(1:read name,2:sequence,3:match stats,4:positions[,])
mCpGbed - encode style mCpG reporting in extended BED format, no auto-detect
(1:chr,2:start,3:end,4:name,5:,6:+/-,7:,8:,9:,10:#C,11:#mC)
allC - Lister style output files detailing the read information about all cytosines
(1:chr,2:pos,3:strand,4:context,#mC,#totalC,#unmC
bismark - Bismark style output files detailing the read information about all cytosines
(1:chr,2:pos,3:strand,4:#mC,5:#unmC,6:context,7:triseq
-minCounts <#> (minimum number of reads to report mC/C ratios, default: 10)
-mCcontext <CG|CHG|CHH|all> (only use C's in this context, default: CG)
HiCsummary - minimal paired-end read mapping information
(1:readname,2:chr1,3:5'pos1,4:strand1,5:chr2,6:5'pos2,7:strand2)
-flip (flip strand of each read, i.e. might want to use with some RNA-seq)
-totalReads <#|all|default> (set the effective total number of reads - all includes multimappers)
-force5th (5th column of BED file contains # of reads mapping to position)
-d <tag directory> [tag directory 2] ... (add Tag directory to new tag directory)
-t <tag file> [tag file 2] ... (add tag file i.e. *.tags.tsv to new tag directory)
-single (Create a single tags.tsv file for all "chromosomes" - i.e. if >100 chromosomes)
-update (Use current tag directory for QC/processing, do not parse new alignment files)
-tbp <#> (Maximum tags per bp, default: no maximum)
-precision <1|2|3> (number of decimal places to use for tag totals, default: 1)
-minlen <#> and -maxlen <#> (Filter reads with lengths outside this range)
GC-bias options:
-genome <genome version> (To see available genomes, use "-genome list")
-or- (for custom genomes):
-genome <path-to-FASTA file or directory of FASTA files>
-checkGC (check Sequence bias, requires "-genome")
-freqStart <#> (offset to start calculating frequency, default: -50)
-freqEnd <#> (distance past fragment length to calculate frequency, default: +50)
-oligoStart <#> (oligo bias start)
-oligoEnd <#> (oligo bias end)
-normGC <target GC profile file> (i.e. tagGCcontent.txt file from control experiment)
Use "-normGC default" to match the genomic GC distribution
-normFixedOligo <oligoFreqFile> (normalize 5' end bias, "-normFixedOligo default" ok)
-minNormRatio <#> (Minimum deflation ratio of tag counts, default: 0.25)
-maxNormRatio <#> (Maximum inflation ratio of tag counts, default: 2.0)
-iterNorm <#> (Sets -max/minNormRatio to 1 and 0, iteratively normalizes such that the
resulting distrubtion is no more than #% different than target, i.e. 0.1,default: off)
-filterReads <seq> <offset> <keep|remove> (filter reads based on oligo sequence in the genome)
HiC options
-removePEbg (remove paired end tags within 1.5x fragment length on same chr)
-PEbgLength <#> (remove PE reads facing on another within this distance, default: 1.5x fragLen)
-restrictionSite <seq> (i.e. AAGCTT for HindIII, assign data < 1.5x fragment length to sites)
Must specify genome sequence directory too. (-rsmis <#> to specify mismatches, def: 0)
-both, -one, -onlyOne, -none (Keeps reads near restriction sites, default: keep all)
-removeSelfLigation (removes reads linking same restriction fragment)
-removeRestrictionEnds (removes reads starting on a restriction fragment)
-assignMidPoint (will place reads in the middle of HindIII fragments)
-restrictionSiteLength <#> (maximum distance from restriction site, default: 1.5x fragLen)
-removeSpikes <size bp> <#> (remove tags from regions with > than # times
the average tags per size bp, suggest "-removeSpikes 10000 8")
-bowtiePE (PE alignments in bowtie alignment, assumes last character of read name is 0 or 1)
(don't need this for sam/bam files)

Can't figure something out? Questions, comments, concerns, or other feedback:
cbenner@ucsd.edu