
HOMER
Software for motif discovery and next-gen sequencing analysis
Retrieving and storing sequencing files
This section will help you figure out where find sequencing
data. Likely places you will find it:- On your lab server/computer (someone already generated
it, or a bioinformatician placed it in a folder for you)
- A scp, ftp, smb, or web server run by your sequencing core or collaborator where your files have been posted
- Public data from GEO/SRA, a lab or company website
Obviously, if you are generating your own sequencing data, talk with your collaborators - they will help you figure out where to find the data.
Accessing private data from a sequencing core
Often a sequencing core will give you a URL or ftp address where you can download your data. Usually this will be password protected, although not always (security though obscurity). Access data this way is usually pretty easy.
The most important files to download are the FASTQ files. These constitute the raw data of the sequencing experiment. More info on FASTQ files is covered in the next section. Most sequencing cores will already demultiplex your samples so that each FASTQ files represents and individual experiment.
It is also worth downloading any instrument files or other quality control statistics. These can be useful if inturment technicians are troubleshooting problematic results.
IMPORTANT: It is important to perform an initial analysis of your data IMMEDIATELY. You may learn quickly that the barcodes used to demultiplex your data were not correct and you lost one of your samples. If you wait too long, the sequencing core may remove your files, so it's best to catch this early so that you can redo the demultiplexing or adjust parameters on the post-sequencing analysis steps that generate the FASTQ files.
After downloading your data, make sure you BACKUP the data on a separate computer in a separate physical location. This may include making a copy on a removable hard drive. The FASTQ files are the raw data, and are needed to publish the experiment. If you loose them you may have trouble publishing your study even if you have downstream analysis files.
Annotation is also important - make sure you label/rename your FASTQ files or place them in directories that makes sense and help you organize your data.
Getting Public Data from GEO/SRA
One very important byproduct of the microarray era was that it is expected that you publish the raw data from high-throughput experiments. This is great because it forces authors to deposit their sequencing data in public repositories. If you are interested in their study, you can download their data and reanalyze their data for your own purposes.
Most data is deposited in NCBI Gene Expression Omnibus (GEO) and/or the NCBI Short Read Archive (SRA). Some data is deposited in the European equivalent ArrayExpress and the European Nucleotide Archive. Japan has their own archive run through the DDBJ.
If you are reading a paper that has high-throughput data, the GEO or SRA should be located near the references. A good strategy is to search for the document for "GSE", which is the first 3 letters of all GEO accession numbers. Sometimes it's found in the methods. Occasionally it's hidden in the supplemental data. If you can't find one, try searching with "SRA" in case it's a SRA accession number.
If you absolutely can't find it, EMAIL the corresponding author, especially if it's a major journal. If they are trying to get away without sharing the data and it's something you're interested in, they need to share it. Ask the author politely for the data in an email. If they are uncooperative, add the journal editor to the email - you'll be surprised how fast that data will appear in GEO if you do that.
Getting the FASTQ files for a sequencing study
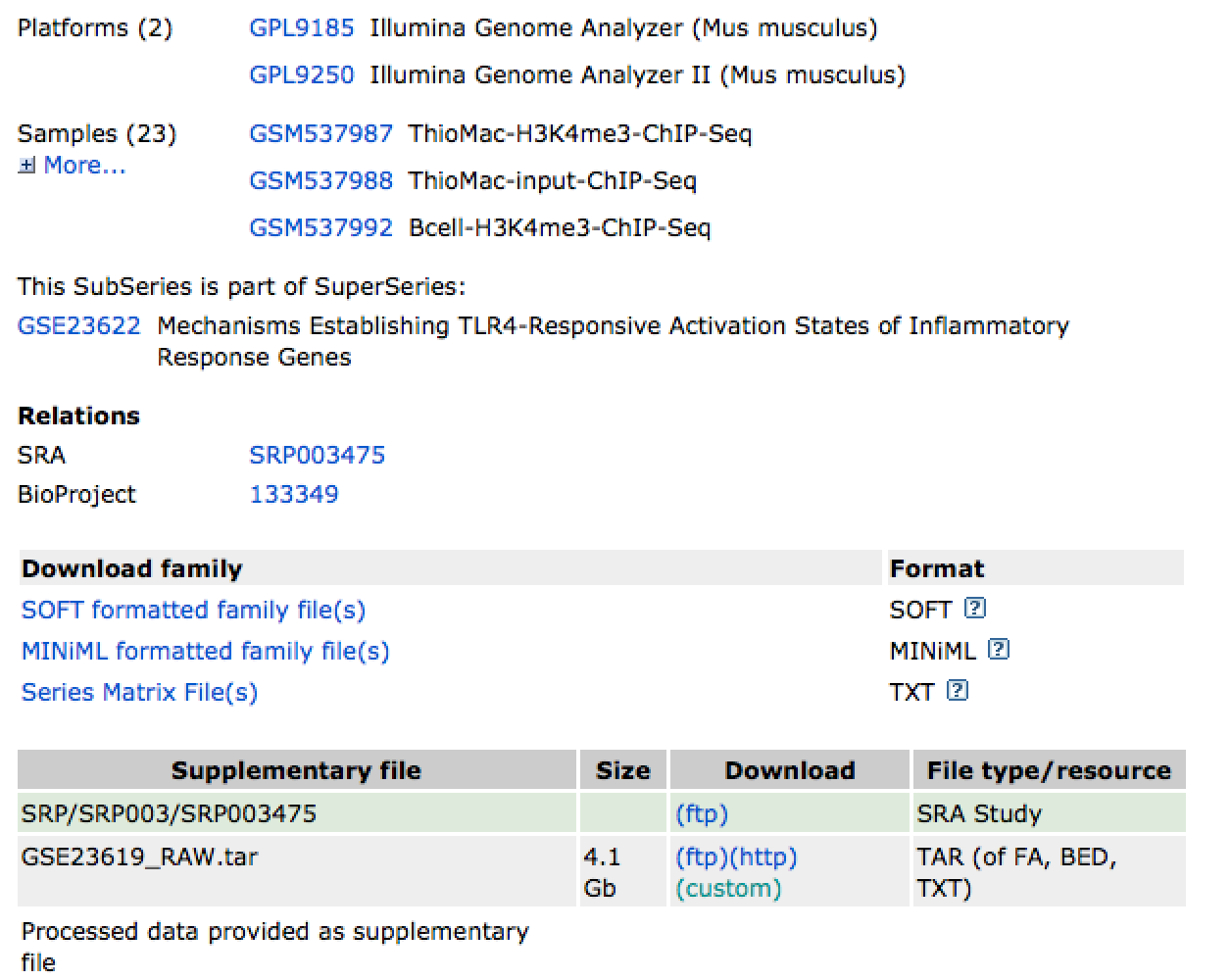
GEO is the most common entry point for sequencing data. Consider the record for GSE23619. The top sections describe the overall experiment, followed by links to individual experiments. Below that are links to the SRA and supplemental files:
To get the RAW sequencing data, you can click on the SRA link, or go to the "Supplementary File" section and click on the (ftp) link to download the data. The easiest way to get the data is to follow the ftp link. This link will lead you to SRA files, such as SRR065223.sra. These are FASTQ files that are specially compressed by the Short Read Archive and need to be opened using a special tool available in the SRA toolkit.
To download and install the SRA toolkit, follow this link and download the appropriate program files. The main program of interest is in the toolkit is called fastq-dump, which is a program that will extract FASTQ files from the SRA files. For example:
fastq-dump SRR065223.sraIf it is a large sequencing study, and you have the tool wget installed, you can download ALL of the SRA files for a study by right clicking on the (ftp) link, and then pasting the URL into the wget command like so:
will produce a file named: SRR065223.fastq
wget -r ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByStudy/sra/SRP%2FSRP003%2FSRP003475/Be sure to add the "-r" which will recursively download the files, and you may also need to add a "/" to the end of the URL to make sure wget knows that you gave it a directory.
After converting files to FASTQ, you should be ready to perform your own analysis on them.
Supplementary Files provided by the author
Sometimes the author provides other useful files, such as sam/bam files (premapped files), peak positions, bedGraph files for visualization, rpkm gene expression counts, etc. Be sure to pay attention to which genome version was used to generate those files.

Can't figure something out? Questions, comments, concerns, or other feedback:
cbenner@salk.edu