
HOMER
Software for motif discovery and next-gen sequencing analysis
Setting up your computing environment
Next-generation sequencing analysis is a compute intensive
process. Your average laptop is probably not up to the
challenge. Most software is geared toward UNIX style
operating systems, with large servers in mind.
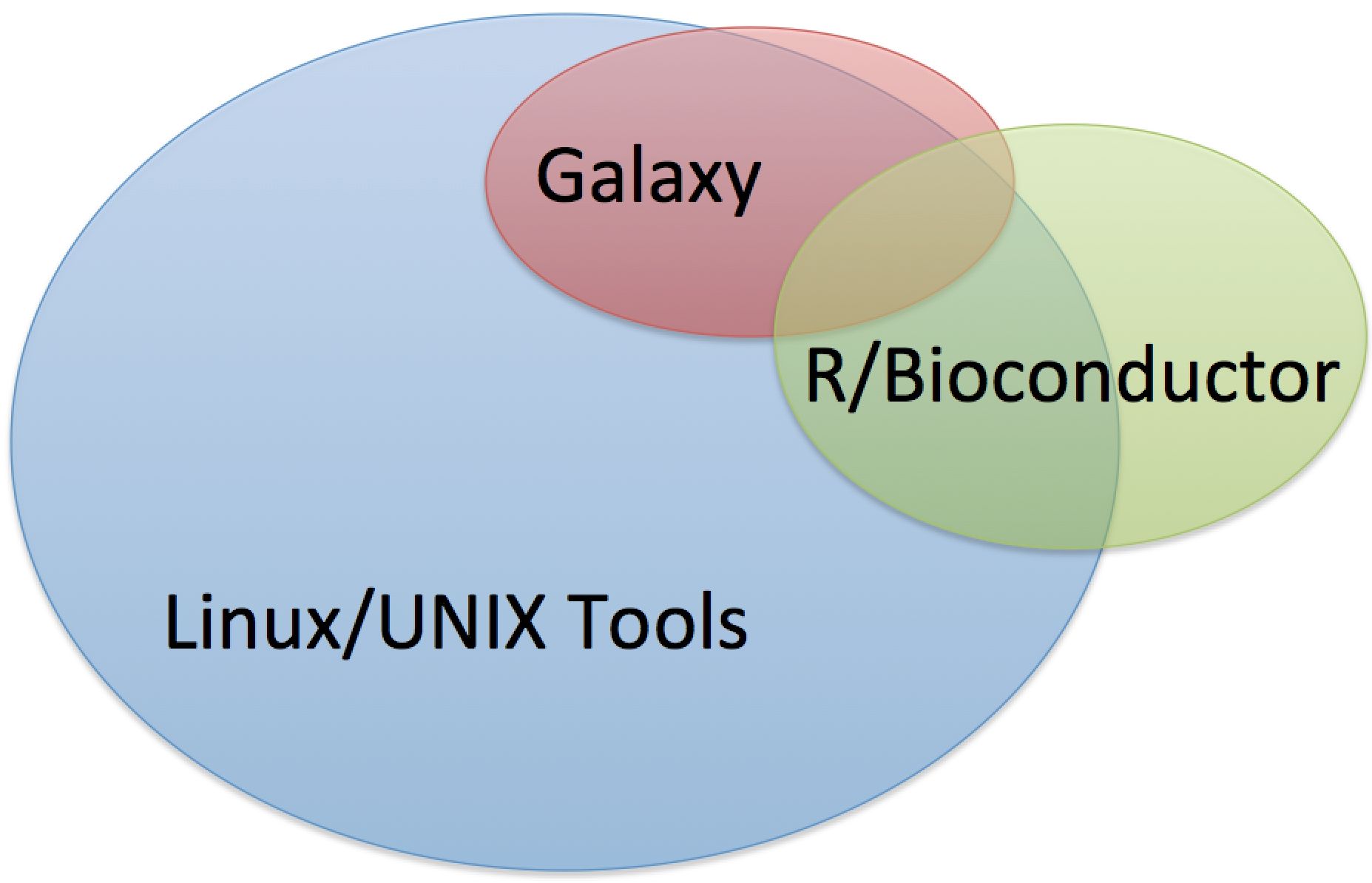
Typically, analysis algorithms will be distributed by
researchers in one of three ways:- Stand alone program to run in a UNIX environment (most common)
- Webserver where you upload data to be analyzed (i.e.
Galaxy)
- R software package/library for the R computing
environment
Some tools are available on multiple platforms, but a vast majority of tools are available as stand alone Linux/UNIX tools.
Types of Software Environments
Linux/UNIX [this tutorial]
Linux, BSD, Solaris, Mac OS X, or really any UNIX-based operating system will work well. Linux systems tend to be the most compatible with academic software, and I find it is easier to install analysis software on Linux than any other operating system. Mac is an attractive choice for many users given it's a good blend between usability and a native UNIX-style operating system. Windows PCs are not ideal given much of the analysis software is not configured to run on them. However, Cygwin is available as a Linux emulator that runs on Windows computers. You can also always consider dual booting your windows system, or using some sort of virtualization software and run Linux at the same time.
The Cloud
Another option is to run your analysis on a web server. These days we call this "analyzing your data in the cloud". By far the most popular "cloud" based analysis tool is Galaxy, which is a compendium of programs (and user/data management tools) for bioinformatics analysis.
This tutorial assumes you will analyze data on your own computer/server. Personally, I feel researchers get more out of it if they learn how to do things on their own, including installing software. Learning things this way also makes you self-reliant. Lets say a new method or new piece of software becomes available to help address a key question with your data. That software is unlikely to be available on Galaxy, and if it does end up being incorporated into Galaxy, it may not end up there for another year. So... you're stuck running it yourself.
R/Bioconductor
Another popular option for next-gen analysis is the statistical computing environment R/Bioconductor(similar to Matlab). R/Bioconductor has a strong community, and R has many built in statistical routines and graphics functions that make developing algorithms in R more attractive than writing your own code in python or C++. Many R programs also run well under Windows, making it a decent option for Windows users
This tutorial will cover the use of a UNIX-style operating system on a local server or individual computer. I feel understanding how to perform analysis from the command line provides researchers with the greatest flexibility, which is key in the fast moving world of next-gen sequencing. For tutorials on how to use Galaxy or R/Bioconductor, visit their respective websites (they are both excellent sources of knowledge).
Computer Hardware
In theory, any computer with enough RAM, hard drive space, and CPU power can be used for analysis - and this includes many laptops. In general, you will need:
4-10 Gb of RAM Minimum (better to have 96+ Gb)For example, if you have a brand new Mac Book Pro laptop with lots of RAM, you can probably do sequencing analysis on it. You may want to sit the computer on a table, because it will get HOT while trying to map 100 million reads to the genome, and might burn your lap if you're sitting with it at home on the couch.
500 Gb of disk space (better to have 10+ Tb of space)
Fast CPU (better to have at least 8 cores, more the better)
Starter Computer: Mac Pro
Optimally, you'll have a dedicated computer/lab server do store data on and perform analysis. This way, you can log on to the server from your personal computer and run analysis remotely. For labs starting out, a popular option is to get a Mac Pro. Not only is it a fast, capable computer, it can double as a shared lab computer that can shared software, like Illustrator.Rack-mount servers
Ideal if you are a lab that does a lot of sequencingSupercomputer Clusters
Often you university or institution provides access to a computing cluster for pennies on the dollar (5-10 cents per CPU hour). These can be great resources for doing genomic mapping if you don't want to invest in more computing power. Usually accessed via UNIX terminal...Virtual/Cloud Computing
Another option (similar to supercomputer clusters) for accessing computing power
Basic Setup of OS/Environment
Linux/UNIX - Not much to do - a basic Linux/UNIX install includes almost all of the basic tools you'll need.
Mac OS - You need to install Xcode, which includes GNU compilers and many other standard UNIX development tools. What sucks is that Xcode is now distributed through the App store, which I hate with a passion. Even though Xcode is free, they'll still basically force you to give them your credit card number... Remember to install the "Command Line tools" package while installing Xcode (you can also install this separately from the Xcode application after installation). I also recommend downloading and installing wget since it's such a useful program after getting Xcode.
Windows - You'll need to install Cygwin. During the Cygwin install, you'll want to install a bunch of the development tools:
Run the install program, and when prompted to choose which packages to install, make sure to install the following:
- GCC compiler, gcc-core, gcc-g++ (in Devel)
- make (in Devel)
- mingw64 (in Devel, for pthread support)
- perl (in perl)
- zip/unzip (in Archive)
- wget (in Web) – very useful for downloading things…
- ghostscript (in Graphics)
Cygwin configuration screen in the setup program (if you already installed cygwin and need to modify it, just rerun the setup program):
Using the Linux/UNIX terminal
Secure Shell (ssh) - accessing remote computers
One of the best features of the UNIX terminal is that it can be used to access remote computer systems. If you have an account on a remote computer and it is running sshd (Secure shell daemon), then you can log onto that computer using ssh client software.
To access a remote computer, you can either use the ssh command from a local terminal (use this if you have Mac OS, Linux, or use Cyginw), or on windows, you may want to use a FREE ssh client program such as PuTTY or select one from here.
To access a remote computer, you must know the hostname or IP address of the computer, and your username/password. To run ssh from the terminal, run the command:
ssh username@hostnameIt will then prompt me for my password, and give me a fresh terminal session on the remote computer.
i.e. ssh cbenner@homer.salk.edu
If you are accessing a server that is always "on", I would highly recommend managing your sessions using screen, which is a persistent terminal session manager that will allow your commands to run even if you log off.
Basic commands:
In UNIX, whenever you type something into the terminal, the first word is considered the "command". Every command works like this:
command [options]Here are some common commands:
cd <directory name> : change your current directory to the directory specified
ls : list the files and directories in your current directory. Add options "-al" to list all details about the files.
pwd : return the full path of your current directory
more <filename> : display the contents of a file
gzip <filename> or gunzip <filename> : GNU zip or unzip a file
tar xvf <filename.tar> : expand a tar archive
tar zxvf <filename.tar.gz> : expand a GNU zipped tar archive. Sometimes these have the file extension *.tgz
top : Useful utility to monitor programs that are currently running.
Getting info about how to run various commands:
<command> --help : will usually return information about how to run the command
man <command> : will display the manual for the command (may not work for every command)
Special keys/commands:
"ctrl+c" : If a command it taking too long, or seems out of control, hit control+C at the same time to quit execution of the current program
<tab>/Autocomplete : Most UNIX terminal programs support auto-complete for file names and program names. While typing a file name in the terminal, if you hit "tab", and there is only one logical choice, the file name will be automatically filled in.
Capturing program output: stdout and stderr
Many UNIX programs will send their output to a special I/O stream called stdout. Normally, stdout is displayed on the screen. However, if you wish to capture this output into a file, you need to add " > filename" to the end of the command. This will capture the output content and save it into the text file "filename".
Understanding the file system
You can think of the UNIX file system as a hierarchical tree. UNIX file systems use a "/" between directory names when specifying a file (windows uses "\"). There are two concepts you must get a handle on when thinking about UNIX filesystems:
To specify the current directory (in a relative sense), use the ".". If you want to specify the parent directory relative to your current position, use "..". This link does an decent job describing how to use file names.
- Absolute File Names (also called the absolute path): Each file has an absolute file name, which starts with a "/". For example, "/home/cbenner/beerRecipe.txt". The absolute file name provides the exact location of the file in the system tree relative to the root (i.e. "/").
- Relative File Names: You have a current location (or present working directory). You can learn what it is using the command "pwd". Any filename that doesn't start with a "/" is a relative file name, and will be describe relative to your current position.
For example, lets say your present working directory is "/home/cbenner". If you specify a file by calling it "data/reads.txt", the system will think you are referring to an absolute file name that is "/home/cbenner/data/reads.txt".
Special directories:
Home Directory: This is the directory where your personal configuration files are stored, and is also usually the first directory that you will be "in" once you log onto the server. There is a special symbol for your home directory: "~/". For example, if your home directory is "/home/chucknorris/", then the file "~/data.txt" is equivalent to "/home/chucknorris/data.txt".File Permissions:
Each file has permissions that allow you to read (e.g. access), write (e.g. modify), and execute (e.g. run the file as a program). These three types of permissions allow you to restrict access to the file. These 3 permissions also apply to yourself, your group, and everyone else on the system. For example if you type "ls -al", you might see something like this:
Chriss-MacBook-Pro:repeats cbenner$ ls -al
total 1152288
drwxr-xr-x 40 cbenner staff 1360 Sep 27 21:47 .
drwxr-xr-x 7 cbenner staff 238 Sep 22 17:28 ..
-rw-r--r-- 1 cbenner staff 84930997 Sep 5 12:44 chimp.L1.txt
-rw-r--r-- 1 cbenner staff 342187 Sep 5 12:44 chimpL1PA2.txt
-rwxr-xr-x 1 cbenner staff 284 Sep 5 21:43 combine.pl
The first several symbols (i.e. drwxr-xr-x) give the permissions on each file. Normally, you don't have to worry much about the permissions until you become comfortable with UNIX, but if you do need to make a program executable, you should run the following command:
chmod 755 program.pl
This will allow you to run the file as a program (i.e. "$ ./program.pl").
Editing text files
In general, there are really only two types of files. ASCII text files, and binary files. ASCII text files are formatted such that each "byte" is represents a character. A byte is composed of a value between 0 and 255, and the meaning of each value is encoded in the ASCII Table. Text files like this are very powerful because virtually any program can interpret them. Most specialized files you may know of are ASCII text files, such as XML, HTML, CSV, and of course TXT. Binary files, on the other hand, can adhere to any format, and are usually meant to be read and written by a single program. An example would be a word doc file, which has a special encoding that is only meant to be accessed my Microsoft Word. For sequencing, a classic example of a text files is a FASTQ file, while a common binary file is a BAM file.
Text files are heavily used in the UNIX world due to their general accessibility. Not only are most data files encoded as text files (like FASTQ), most UNIX configuration files are also text files. There are several common utilities included in UNIX to manipulate text files, such as more, cut, wc, cat, etc.
Text Editors:
Knowing how to use UNIX Text Editors is important when achieving comfort with UNIX. Below are three common editors, with links to pages the give some background information about how to use them. Each will have countless tutorials covering their use (just google "<editor> tutorial":
vi : the most common UNIX text editor, found on any system. Very powerful, but unfortunately a little difficult to use for beginners. Worth learning, but takes some effort.
pico : one of the easiest UNIX text editors to use, good for beginners, not as popular as vi.
emacs : one of the most extensible UNIX editors, common for power-users.
Installing Software
There are two general approaches to installing software on a Linux/UNIX system. The first is to use a package manager (like yum or apt-get). The second is to directly download the software and install it from source (or simply download the executable program).
Package Managers:
Installing software with package managers is great because it will make sure all "prerequisites" are also installed to make sure your software runs as planned. This is typically only an option for well known programs (most academic software must be installed directly from source).Installing software from source:
If running a debian flavor of Linux (i.e. Ubuntu), then you will use something like this:
apt-get install <package name>If running a redhat flavor of Linux (i.e. CentOS), then you will use a different package manager, such as:
yum install <package name>In many cases, you must be the root (or superuser) to install system software, so you may have to add "sudo" to the beginning of these commands.
Most academic software must be directly downloaded from the author's website. Software like this can come in two flavors, it can be source files or it can be pre-compiled/binary files (programs ready to run). If you are downloading programs that are pre-compiled, make sure you get the files specific to your system (i.e. Mac OSX, Linux x86_64, Solaris, etc.)
It might sound daunting to install software from source, but usually it is not bad at all. In 90% of cases, you'll simply run the next couple commands:
- Download the software to your current directory
- type: "tar zxvf program.tar.gz"
- This will likely make a new directory, use "cd programDirectory" to move into the new directory.
- Read the "README.txt" and "INSTALL.txt" files, if they exist. These files should include directions about how to install the software.
- Usually, this means running the next three commands:
./configure
make
sudo make install
Changing your executable PATH variable
Every program in UNIX is actually a file. Most program files for system commands, like "cd" or "ls" are found in special directories such as "/usr/bin" or "/usr/local/bin". Whenever you type something into the terminal, the first word you type is the command. When you press enter, the operating system attempt to find the program file corresponding to the command. For example, if you type "ls", it needs to find the file with the execution instructions that will list your files.
The location of program files is managed in UNIX systems using the PATH variable. This is a variable maintained by the shell (e.g. terminal program) that stores the directories that contain program files. If you run the command "echo $PATH", the contents of the PATH variable will be shown:
echo $PATH
/usr/lib64/qt-3.3/bin:/usr/local/bin:/bin:/usr/bin:/usr/local/sbin:/usr/sbin:/sbin:/bioinf/seqlogo/weblogo:/bioinf/ucsc:/bioinf/homer/bin/:/bioinf/samtools/samtools-0.1.18/
What is shown is a list of directories separated by colons ":". When you type a command, the operating system will go to each of these directories (in order) and look for an executable program file matching the name of the one you typed. If you want to figure out where the command is, you can use the "which <command>" command to return the location of the command you ran. For example:
which ls
/bin/ls
This shows that the ls command is actually a program file located in the /bin/ directory.
Why is this important? Often, you may download a program and put it in a directory - lets say I download a program called "roundhouse" and place it in the directory "/home/chucknorris/software/". I then make sure it is executable by changing it's permissions: "chmod 755 roundhouse". To run the program from anywhere, I must specify the program file location explicitly: "/home/chucknorris/software/roundhouse". However, if I want to run it like most commands (i.e. just type "roundhouse"), I need to place it in a directory that the operating system will search for (e.g. a directory that is included with the PATH variable).
For that, you have two options. First, you could copy the roundhouse program to a directory that is in the PATH, such as copying it to the /bin/ directory. However, this is usually not a good idea since the bin directory is normally reserved for system applications. Another approach is to modify your PATH variable to include the /home/chucknorris/software/ directory so that the operating system will search it when looking for program files.
The best way to do this (assuming you are using the bash shell, which is the most common), is to edit the "~/.bashrc" resource file. Using vi or pico, edit the file and include a line such as this one:
PATH=$PATH:/hoem/chucknorris/software
The "~/.bashrc" file is a special resource file that is executed whenever you login. This way, whenever you log into your computer, your PATH variable will be updated to include the special directory.

Can't figure something out? Questions, comments, concerns, or other feedback:
cbenner@salk.edu